

1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway
- PMID: 26287999
- PMCID: PMC4646414
- DOI: 10.1097/MIB.0000000000000526
1,25-Dihydroxyvitamin D Protects Intestinal Epithelial Barrier by Regulating the Myosin Light Chain Kinase Signaling Pathway
Abstract
Background: The myosin light chain kinase (MLCK) pathway controls intestinal epithelial barrier permeability by regulating the tight junction. 1,25-dihydroxyvitamin D (1,25(OH)2D3)-vitamin D receptor (VDR) signaling protects the epithelial barrier, but the molecular mechanism is incompletely understood.
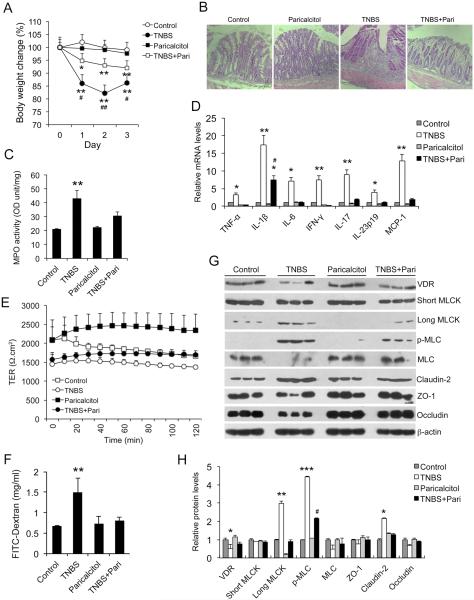
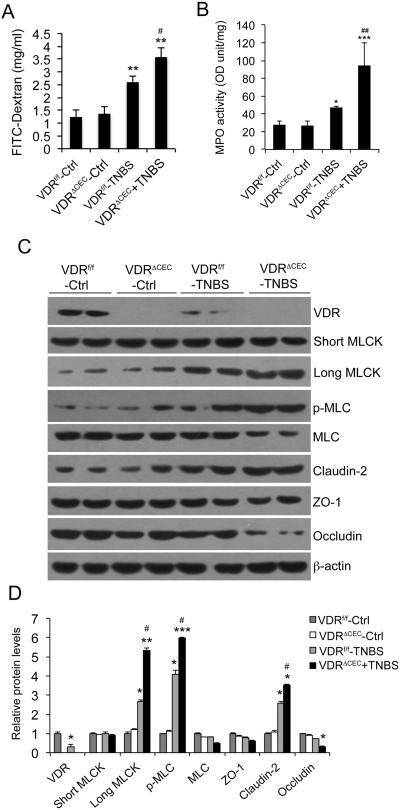
Methods: MLCK activation and barrier permeability were studied using monolayers of HCT116, Caco-2, and SW480 cells treated with tissue necrosis factor α with or without 1,25(OH)2D3. The MLCK pathway was analyzed in normal and inflamed colonic biopsies from patients with ulcerative colitis. Colonic mucosal barrier permeability and MLCK activation were also investigated using trinitrobenzene sulfonic acid-induced colitis models in vitamin D analog paricalcitol-treated wild-type mice and mice carrying VDR deletion in colonic epithelial cells.
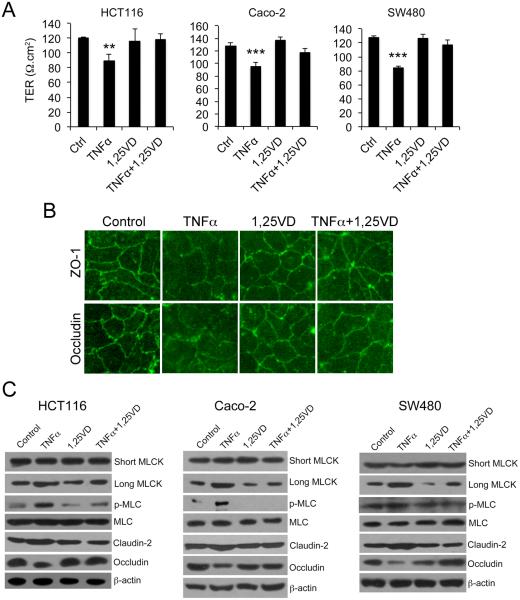
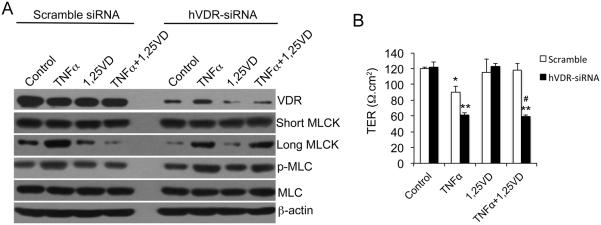
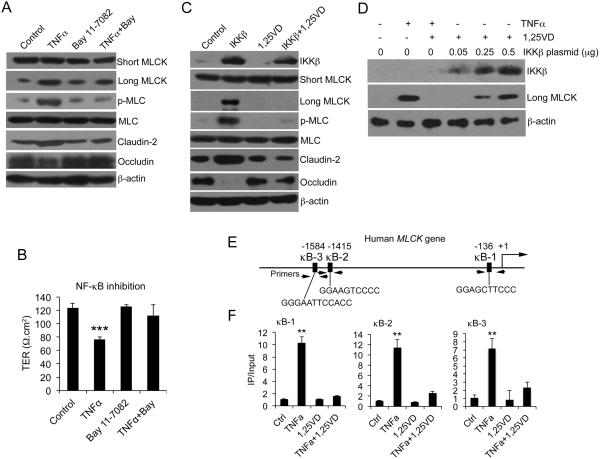
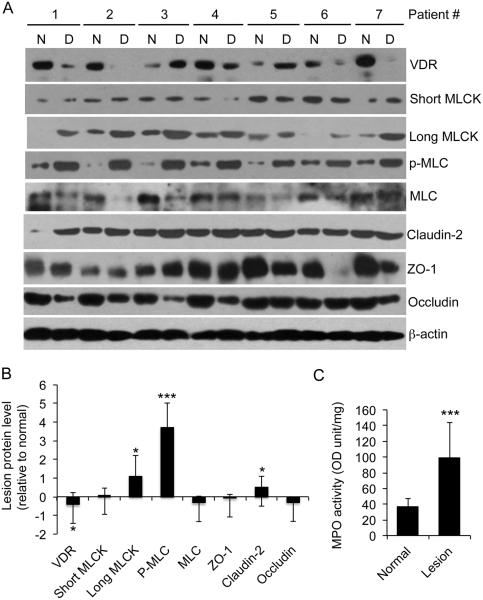
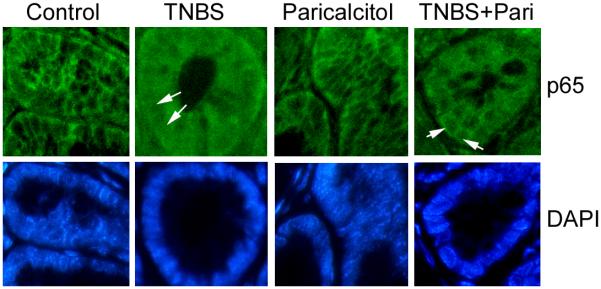
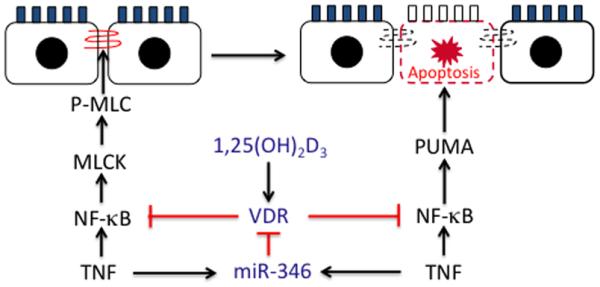
Results: Tissue necrosis factor α increased cell monolayer permeability and induced long isoform of MLCK expression and myosin II regulatory light chain (MLC) phosphorylation, and 1,25(OH)2D3 blocked tissue necrosis factor α-induced increases in monolayer permeability and MLCK-MLC pathway activation by a VDR-dependent fashion. 1,25(OH)2D3 directly suppressed long MLCK expression by attenuating NF-κB activation, and chromatin immunoprecipitation assays confirmed that 1,25(OH)2D3 disrupted p65 binding to 3 κB sites in long MLCK gene promoter. In human ulcerative colitis biopsies, VDR reduction was associated with increases in long MLCK expression and MLC phosphorylation. In trinitrobenzene sulfonic acid colitis models, paricalcitol ameliorated colitis, attenuated the increase in mucosal barrier permeability, and inhibited long MLCK induction and MLC phosphorylation. In contrast, mice with colonic epithelial VDR deletion exhibited more robust increases in mucosal barrier permeability and MLCK activation compared with wild-type mice.
Conclusions: These data demonstrate that 1,25(OH)2D3-VDR signaling preserves the mucosal barrier integrity by abrogating MLCK-dependent tight junction dysregulation during colonic inflammation.
Figures
References
-
- Laukoetter MG, Bruewer M, Nusrat A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr Opin Gastroenterol. 2006;22:85–89. - PubMed
-
- Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:416–422. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
