

SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures
- PMID: 26291284
- PMCID: PMC4567464
- DOI: 10.1212/WNL.0000000000001926
SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures
Abstract
Objective: De novo SCN2A mutations have recently been associated with severe infantile-onset epilepsies. Herein, we define the phenotypic spectrum of SCN2A encephalopathy.
Methods: Twelve patients with an SCN2A epileptic encephalopathy underwent electroclinical phenotyping.
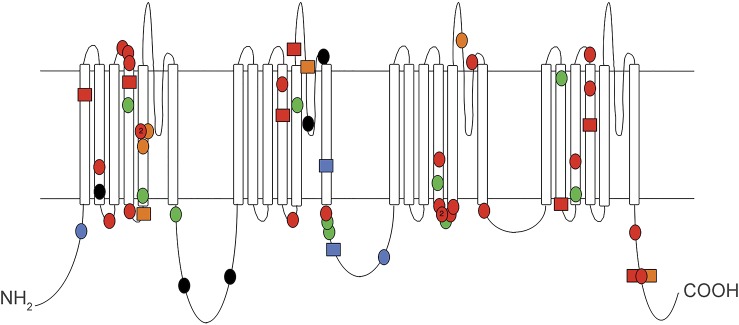
Results: Patients were aged 0.7 to 22 years; 3 were deceased. Seizures commenced on day 1-4 in 8, week 2-6 in 2, and after 1 year in 2. Characteristic features included clusters of brief focal seizures with multiple hourly (9 patients), multiple daily (2), or multiple weekly (1) seizures, peaking at maximal frequency within 3 months of onset. Multifocal interictal epileptiform discharges were seen in all. Three of 12 patients had infantile spasms. The epileptic syndrome at presentation was epilepsy of infancy with migrating focal seizures (EIMFS) in 7 and Ohtahara syndrome in 2. Nine patients had improved seizure control with sodium channel blockers including supratherapeutic or high therapeutic phenytoin levels in 5. Eight had severe to profound developmental impairment. Other features included movement disorders (10), axial hypotonia (11) with intermittent or persistent appendicular spasticity, early handedness, and severe gastrointestinal symptoms. Mutations arose de novo in 11 patients; paternal DNA was unavailable in one.
Conclusions: Review of our 12 and 34 other reported cases of SCN2A encephalopathy suggests 3 phenotypes: neonatal-infantile-onset groups with severe and intermediate outcomes, and a childhood-onset group. Here, we show that SCN2A is the second most common cause of EIMFS and, importantly, does not always have a poor developmental outcome. Sodium channel blockers, particularly phenytoin, may improve seizure control.
© 2015 American Academy of Neurology.
Figures



References
-
- Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia 2012;53:1849–1859. - PubMed
-
- Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet 2002;360:851–852. - PubMed
-
- Berkovic SF, Heron SE, Giordano L, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol 2004;55:550–557. - PubMed
-
- Shi X, Yasumoto S, Nakagawa E, et al. Missense mutation of the sodium channel gene SCN2A causes Dravet syndrome. Brain Dev 2009;31:758–762. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases