

Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies
- PMID: 26295289
- PMCID: PMC4594321
- DOI: 10.17305/bjbms.2015.636
Dystrophin and the two related genetic diseases, Duchenne and Becker muscular dystrophies
Abstract
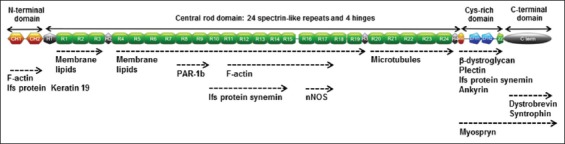
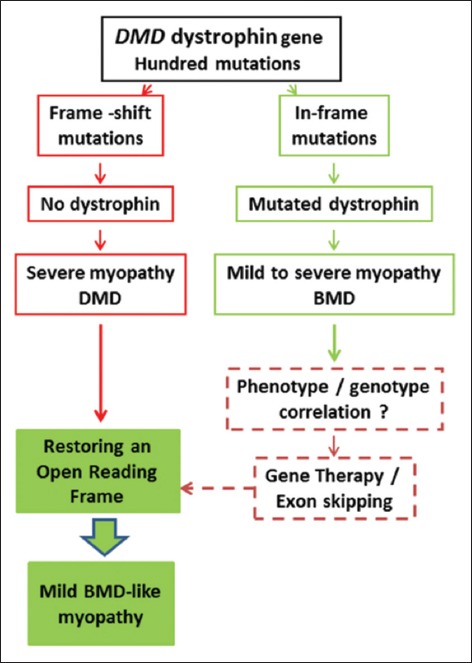
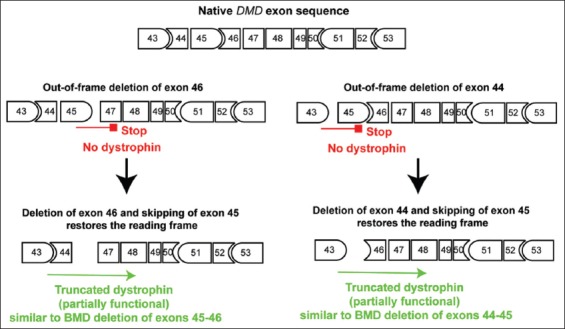
Mutations of the dystrophin DMD gene, essentially deletions of one or several exons, are the cause of two devastating and to date incurable diseases, Duchenne (DMD) and Becker (BMD) muscular dystrophies. Depending upon the preservation or not of the reading frame, dystrophin is completely absent in DMD, or present in either a mutated or a truncated form in BMD. DMD is a severe disease which leads to a premature death of the patients. Therapy approaches are evolving with the aim to transform the severe DMD in the BMD form of the disease by restoring the expression of a mutated or truncated dystrophin. These therapies are based on the assumption that BMD is a mild disease. However, this is not completely true as BMD patients are more or less severely affected and no molecular basis of this heterogeneity of the BMD form of the disease is yet understood. The aim of this review is to report for the correlation between dystrophin structures in BMD deletions in view of this heterogeneity and to emphasize that examining BMD patients in details is highly relevant to anticipate for DMD therapy effects.
Figures


References
-
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987 Jul 31;50:509–17. http://dx.doi.org/10.1016/0092-8674(87)90504-6 . - PubMed
-
- Kenwrick S, Patterson M, Speer A, Fischbeck K, Davies K. Molecular analysis of the Duchenne muscular dystrophy region using pulsed field gel electrophoresis. Cell. 1987 Jan 30;48:351–7. http://dx.doi.org/10.1016/0092-8674(87)90438-7 . - PubMed
-
- Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009 Dec;30:1657–66. http://dx.doi.org/10.1002/humu.21114 . - PMC - PubMed
-
- Tuffery-Giraud S, Beroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009 Jun;30:934–45. http://dx.doi.org/10.1002/humu.20976 . - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
