

Short-term high glucose exposure impairs insulin signaling in endothelial cells
- PMID: 26297582
- PMCID: PMC4546318
- DOI: 10.1186/s12933-015-0278-0
Short-term high glucose exposure impairs insulin signaling in endothelial cells
Abstract
Background: Hyperglycemia is the hallmark of diabetes and its cardiovascular complications. Insulin plays an important role in the regulation of vascular homeostasis and maintenance of endothelial function. Insulin signaling occurs after binding to the insulin receptor, causing activation of two separate and parallel pathways: PI3K/AKT/eNOS and Ras/Raf/MAPK pathways. AKT phosphorylates eNOS at Ser1177, resulting in increased nitric oxide production and vasodilation. The MAPK pathway results in endothelin-1 production and vasoconstriction and mitogenic effects.
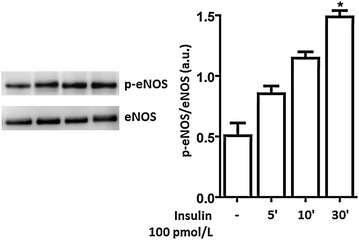
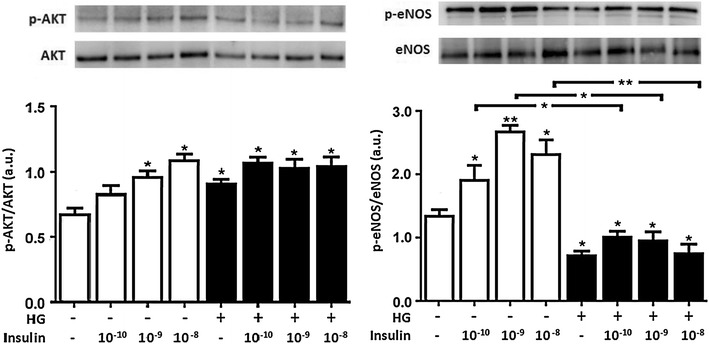
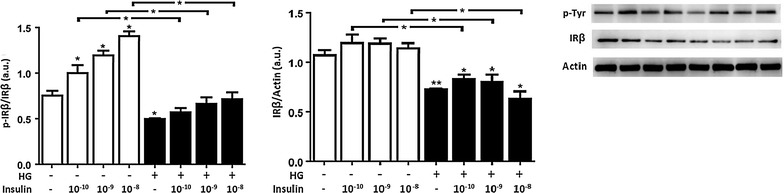
Methods: We studied the effects of physiological insulin treatment in human umbilical vein endothelial cells (HUVECs) on the two pathways under high glucose conditions, which mimic the in vivo condition of hyperglycemia. HUVECs were incubated with insulin at different physiological concentrations (from 10(-10) to 10(-8) M) for 30 min after 24 h of exposition to normal (5 mmol/L, NG) or high glucose (25 mmol/L, HG). Phosphorylated forms of AKT, eNOS, ERK1/2, p38, JNK and insulin receptor-β subunit (IRβ) were evaluated.
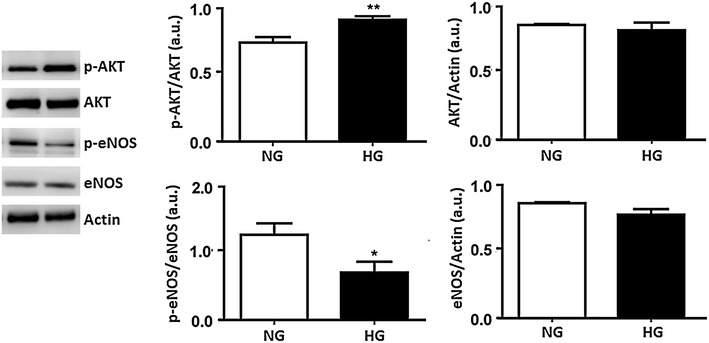
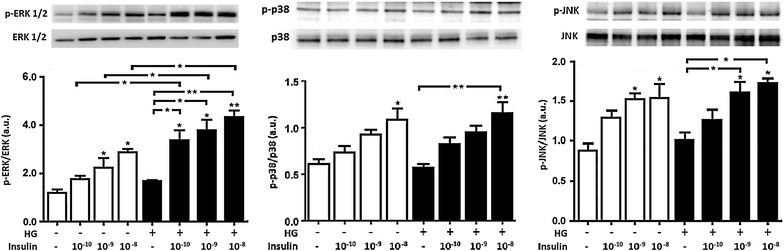
Results: In normal glucose, the active phosphorylated forms of AKT, eNOS, ERK1/2, p38 and JNK were increased in insulin treated cells, in a dose-dependent manner. In high glucose, insulin was not able to activate the PI3K/AKT/eNOS pathway, with the phosphorylated form of eNOS reduced with respect to the control. However, insulin was able to induce the up-regulation of phospho-ERK1/2, -p38 and -JNK. Moreover, we found reduced levels of IRβ phosphorylated form in high glucose as compared to the control. Insulin was able to increase phospho-IRβ in normal glucose but not in high glucose, in which the total protein levels remained reduced.
Conclusions: Exposure to short-term high glucose negatively affects insulin signaling even when physiological insulin concentrations are added. The impairment of the PI3K/AKT/eNOS pathway after physiological insulin treatment could contribute to detrimental effects on cardiovascular homeostasis under high glucose conditions, and might shift toward the activation of certain mitogenic effectors, such as ERK1/2, p38 and JNK, the only ones that respond to physiological insulin treatment in high glucose.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
