

Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome
- PMID: 26299364
- PMCID: PMC4564989
- DOI: 10.1016/j.ajhg.2015.07.015
Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome
Abstract
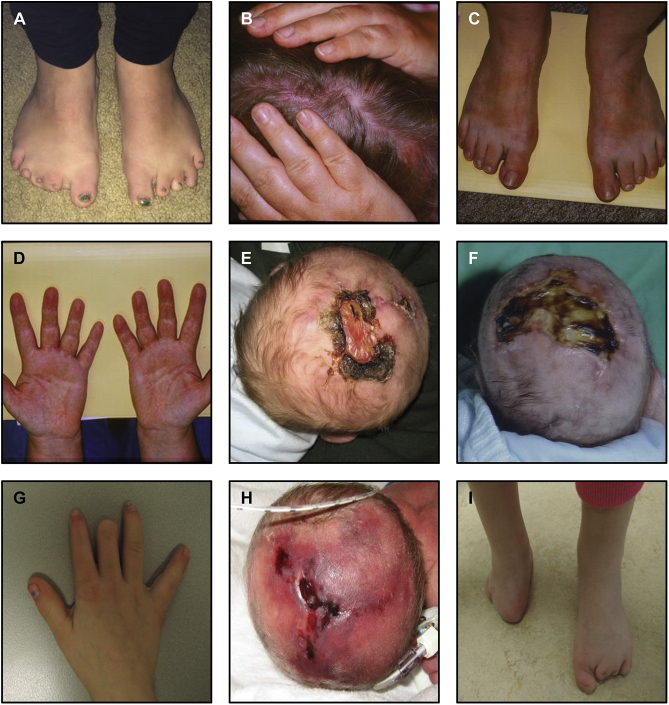
Adams-Oliver syndrome (AOS) is a rare developmental disorder characterized by the presence of aplasia cutis congenita (ACC) of the scalp vertex and terminal limb-reduction defects. Cardiovascular anomalies are also frequently observed. Mutations in five genes have been identified as a cause for AOS prior to this report. Mutations in EOGT and DOCK6 cause autosomal-recessive AOS, whereas mutations in ARHGAP31, RBPJ, and NOTCH1 lead to autosomal-dominant AOS. Because RBPJ, NOTCH1, and EOGT are involved in NOTCH signaling, we hypothesized that mutations in other genes involved in this pathway might also be implicated in AOS pathogenesis. Using a candidate-gene-based approach, we prioritized DLL4, a critical NOTCH ligand, due to its essential role in vascular development in the context of cardiovascular features in AOS-affected individuals. Targeted resequencing of the DLL4 gene with a custom enrichment panel in 89 independent families resulted in the identification of seven mutations. A defect in DLL4 was also detected in two families via whole-exome or genome sequencing. In total, nine heterozygous mutations in DLL4 were identified, including two nonsense and seven missense variants, the latter encompassing four mutations that replace or create cysteine residues, which are most likely critical for maintaining structural integrity of the protein. Affected individuals with DLL4 mutations present with variable clinical expression with no emerging genotype-phenotype correlations. Our findings demonstrate that DLL4 mutations are an additional cause of autosomal-dominant AOS or isolated ACC and provide further evidence for a key role of NOTCH signaling in the etiology of this disorder.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

Comment in
-
DLL4 loss-of-function heterozygous mutations cause Adams-Oliver syndrome.Clin Genet. 2015 Dec;88(6):532. doi: 10.1111/cge.12681. Epub 2015 Oct 16. Clin Genet. 2015. PMID: 26419402 No abstract available.
References
-
- Forrest H., Adams C.P.O. Hereditary deformities in man due to arrested development. J. Hered. 1945;36:3–7.
-
- Snape K.M., Ruddy D., Zenker M., Wuyts W., Whiteford M., Johnson D., Lam W., Trembath R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A. 2009;149A:1860–1881. - PubMed
-
- Shaheen R., Faqeih E., Sunker A., Morsy H., Al-Sheddi T., Shamseldin H.E., Adly N., Hashem M., Alkuraya F.S. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am. J. Hum. Genet. 2011;89:328–333. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
