

RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer
- PMID: 26301689
- PMCID: PMC4734742
- DOI: 10.1038/nm.3930
RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer
Abstract
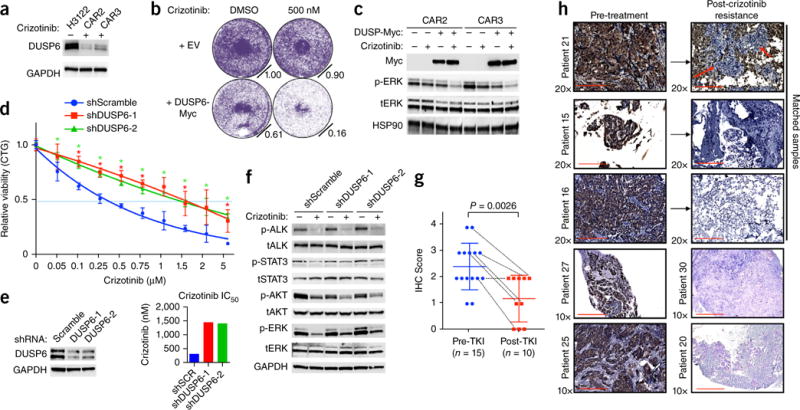
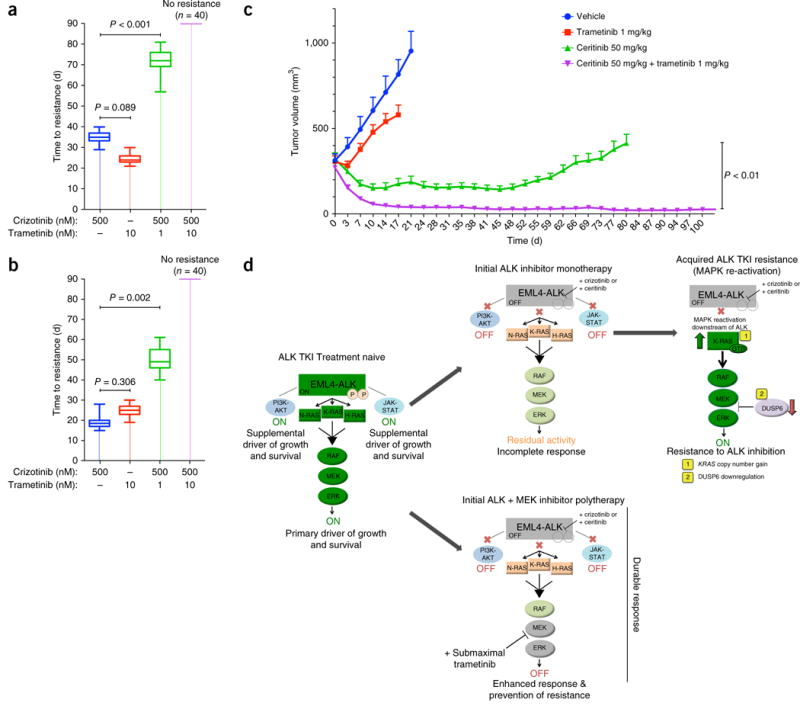
One strategy for combating cancer-drug resistance is to deploy rational polytherapy up front that suppresses the survival and emergence of resistant tumor cells. Here we demonstrate in models of lung adenocarcinoma harboring the oncogenic fusion of ALK and EML4 that the GTPase RAS-mitogen-activated protein kinase (MAPK) pathway, but not other known ALK effectors, is required for tumor-cell survival. EML4-ALK activated RAS-MAPK signaling by engaging all three major RAS isoforms through the HELP domain of EML4. Reactivation of the MAPK pathway via either a gain in the number of copies of the gene encoding wild-type K-RAS (KRAS(WT)) or decreased expression of the MAPK phosphatase DUSP6 promoted resistance to ALK inhibitors in vitro, and each was associated with resistance to ALK inhibitors in individuals with EML4-ALK-positive lung adenocarcinoma. Upfront inhibition of both ALK and the kinase MEK enhanced both the magnitude and duration of the initial response in preclinical models of EML4-ALK lung adenocarcinoma. Our findings identify RAS-MAPK dependence as a hallmark of EML4-ALK lung adenocarcinoma and provide a rationale for the upfront inhibition of both ALK and MEK to forestall resistance and improve patient outcomes.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper.
Figures
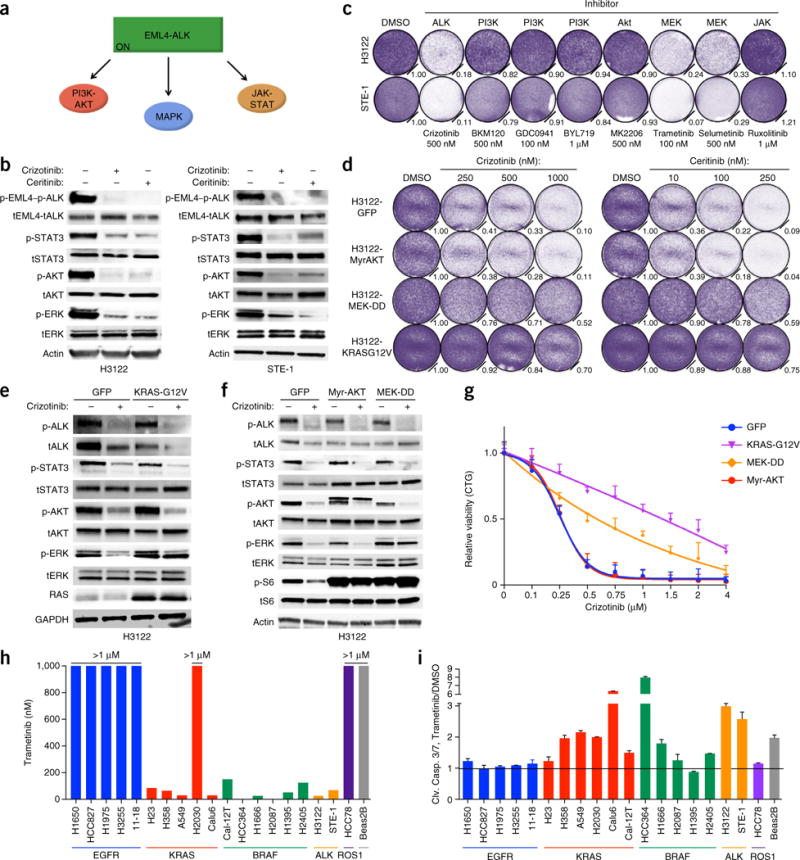
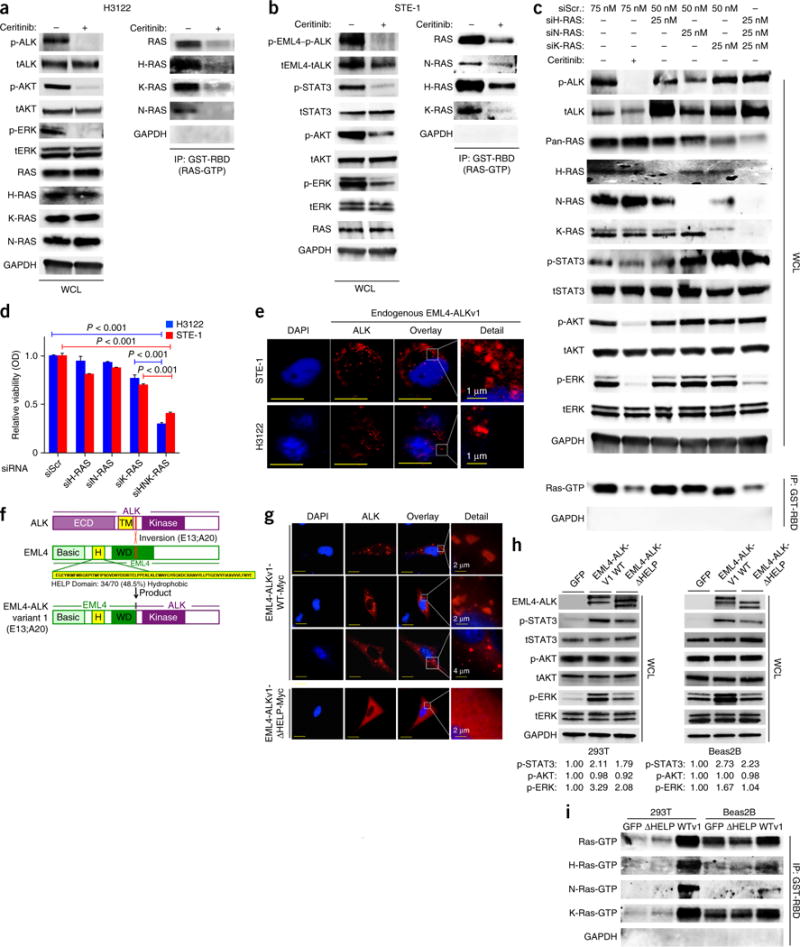
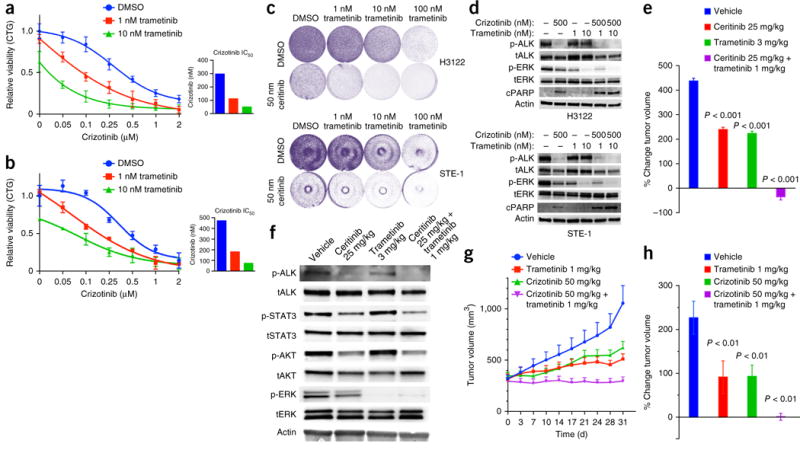
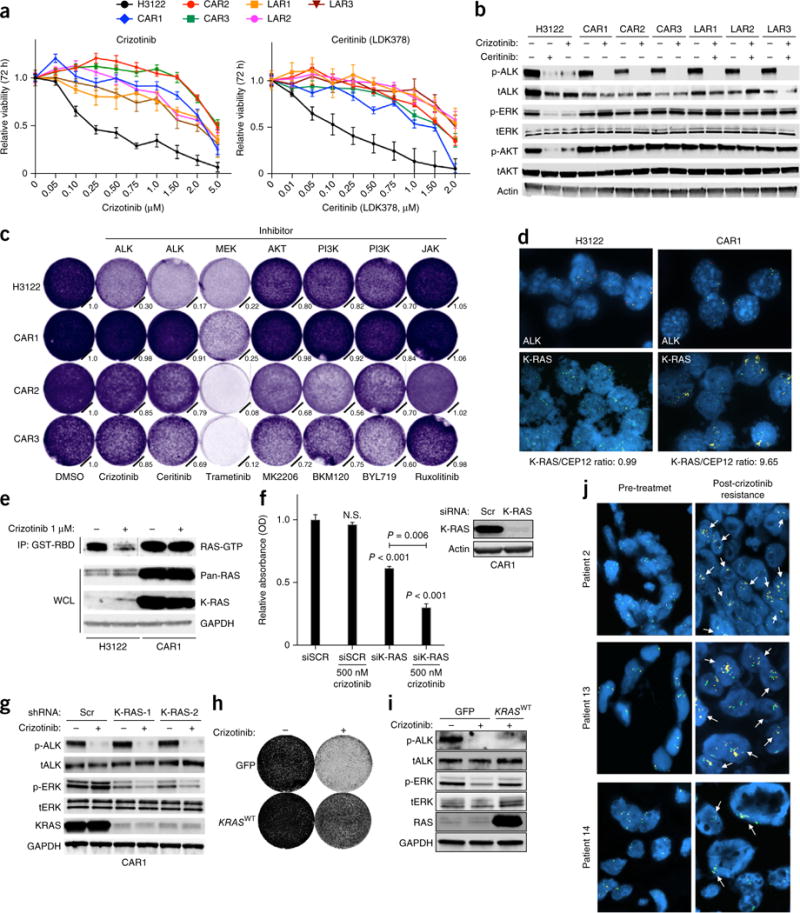
Comment in
-
Up-front polytherapy for ALK-positive lung cancer.Nat Med. 2015 Sep;21(9):974-5. doi: 10.1038/nm.3942. Nat Med. 2015. PMID: 26340116 No abstract available.
References
-
- Solomon BJ, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–2177. - PubMed
-
- Shaw AT, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. - PubMed
-
- Robert C, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P50 CA058187/CA/NCI NIH HHS/United States
- P30CA046934/CA/NCI NIH HHS/United States
- R01 CA211052/CA/NCI NIH HHS/United States
- P30 CA046934/CA/NCI NIH HHS/United States
- R01 CA121210/CA/NCI NIH HHS/United States
- DP2 CA174497/CA/NCI NIH HHS/United States
- K12 CA138464/CA/NCI NIH HHS/United States
- 5K12CA086913/CA/NCI NIH HHS/United States
- K12 CA086913/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- P50CA058187/CA/NCI NIH HHS/United States
- P01CA129243/CA/NCI NIH HHS/United States
- R01 CA169338/CA/NCI NIH HHS/United States
- R01CA121210/CA/NCI NIH HHS/United States
- T32 CA128583/CA/NCI NIH HHS/United States
- P01 CA129243/CA/NCI NIH HHS/United States
- K12CA138464/CA/NCI NIH HHS/United States
- K08 CA154787/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
