

doi: 10.1038/nmeth.3547.
Epub 2015 Aug 24.
Predicting effects of noncoding variants with deep learning-based sequence model
Affiliations
- PMID: 26301843
- PMCID: PMC4768299
- DOI: 10.1038/nmeth.3547
Item in Clipboard
Predicting effects of noncoding variants with deep learning-based sequence model
Nat Methods.
2015 Oct.
Abstract
Identifying functional effects of noncoding variants is a major challenge in human genetics. To predict the noncoding-variant effects de novo from sequence, we developed a deep learning-based algorithmic framework, DeepSEA (http://deepsea.princeton.edu/), that directly learns a regulatory sequence code from large-scale chromatin-profiling data, enabling prediction of chromatin effects of sequence alterations with single-nucleotide sensitivity. We further used this capability to improve prioritization of functional variants including expression quantitative trait loci (eQTLs) and disease-associated variants.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
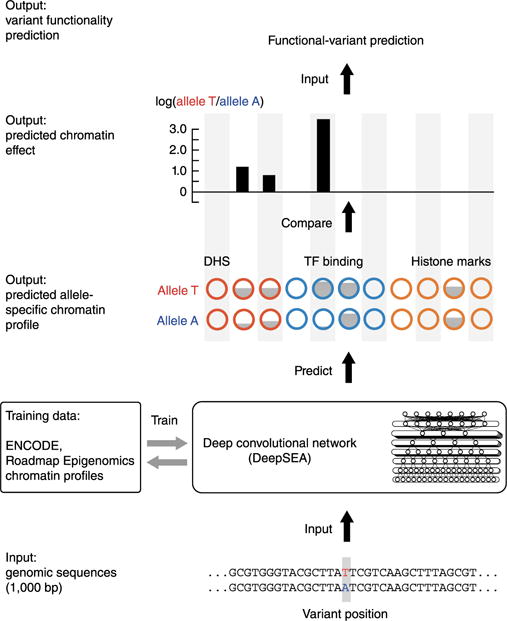
Schematic overview of the DeepSEA pipeline, a strategy for predicting chromatin effects of noncoding variants.
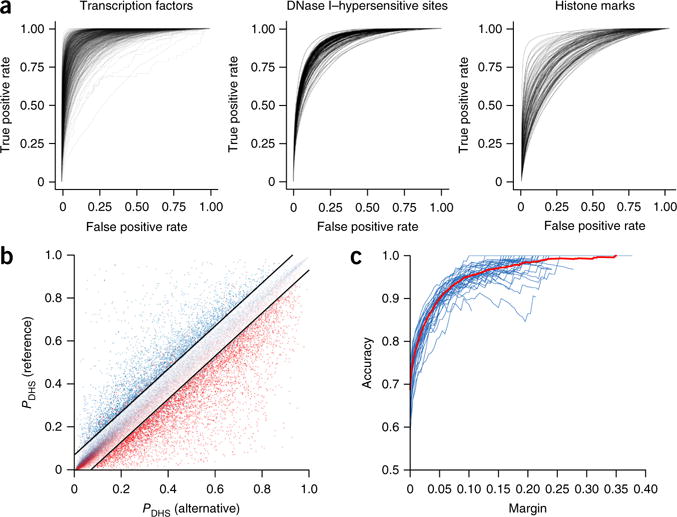
The deep-learning model accurately predicts chromatin features from sequence with single-nucleotide sensitivity. (a) Receiver operating characteristic (ROC) curves for each TF (left), DNase-seq (center) and histone-mark (right) profile prediction. Chromatin features with at least 50 test-positive samples were used. (b) DeepSEA predictions for DNase I–sensitive alleles of 57,407 allelically imbalanced variants from the digital genomic footprinting (DGF) DNase-seq data for 35 different cell types. The y and x axes show, respectively, for a variant, the predicted probabilities that the sequences carrying the reference allele and the alternative allele are DHSs within the corresponding cell type. The red and blue dots represent, respectively, the experimentally determined alternative allele–biased and reference allele–biased variants as determined by DGF data. The black lines indicate the margin, or the threshold of predicted probability differences between the two alleles for classifying high-confidence predictions (margin = 0.07 for this plot). (c) Accuracy. Each blue line indicates the performance for a different cell type, and the red line shows the overall performance on allelically imbalanced variants for all 35 cell types.
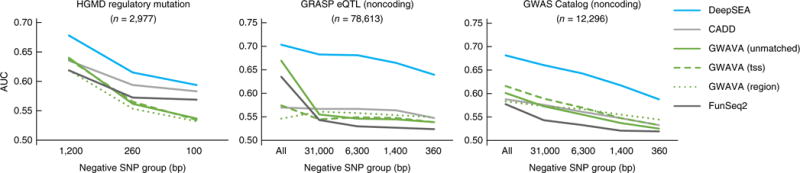
Sequence-based prioritization of functional noncoding variants. Comparison of DeepSEA to other methods for prioritizing functionally annotated variants including HGMD annotated regulatory mutations, noncoding GRASP eQTLs and noncoding GWAS Catalog SNPs against noncoding 1000 Genomes Project SNPs (across multiple negative-variant groups with different scales of distances to the positive SNPs). The x axes show the average distances of negative-variant groups to a nearest positive variant. The “All” negative-variant groups are randomly selected negative 1000 Genomes SNPs. Because GWAVA was trained on the HGMD regulatory mutations, we filtered out GWAVA training positive-variant examples and closely located variants (within 2,000 bp) in evaluating its performance on HGMD regulatory mutations. Model performance is measured with area under the receiver operating characteristic curves (AUC).
Comment in
-
Diving deeper to predict noncoding sequence function.Nat Methods. 2015 Oct;12(10):925-6. doi: 10.1038/nmeth.3604. Nat Methods. 2015. PMID: 26418766 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
