

Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape
- PMID: 26304884
- PMCID: PMC4567701
- DOI: 10.1200/JCO.2014.59.9217
Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape
Abstract
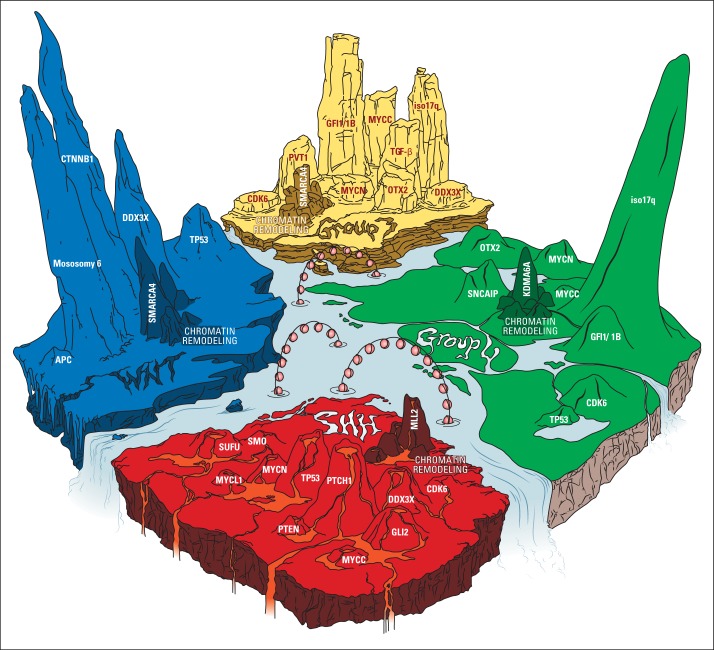
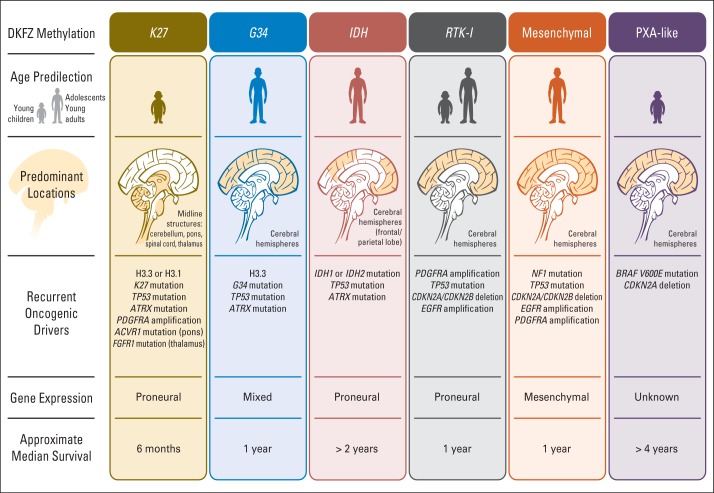
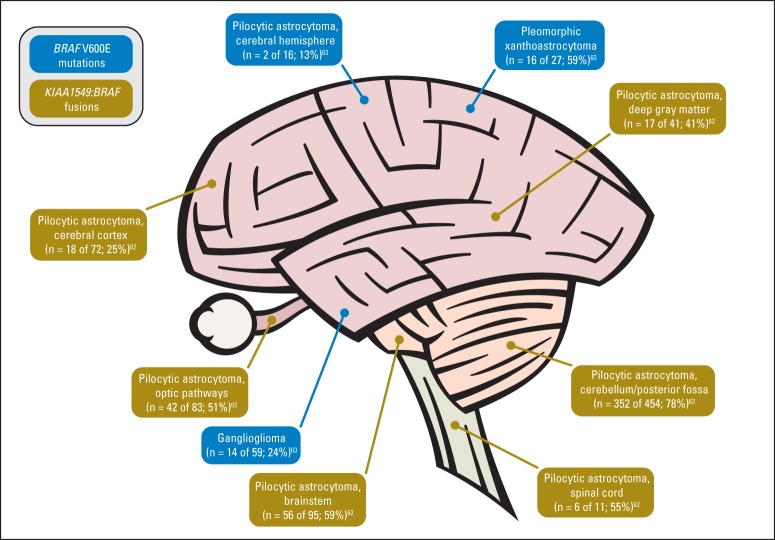
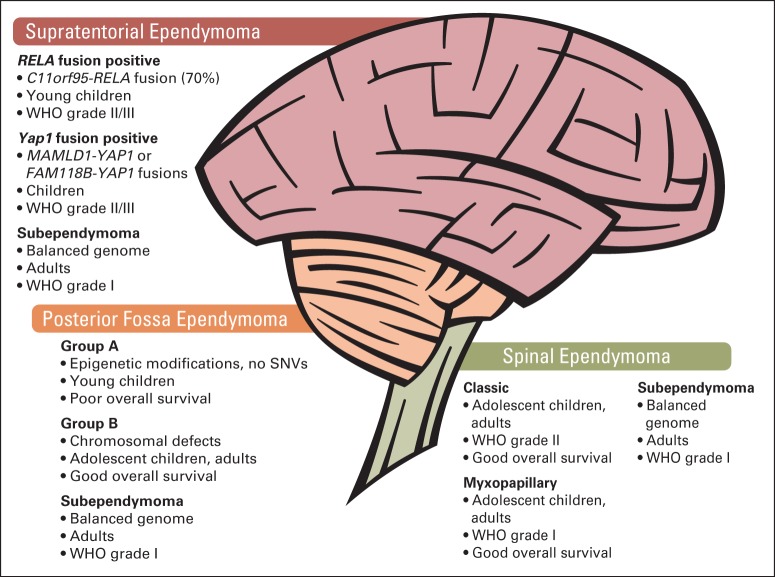
Pediatric neuro-oncology has undergone an exciting and dramatic transformation during the past 5 years. This article summarizes data from collaborative group and institutional trials that have advanced the science of pediatric brain tumors and survival of patients with these tumors. Advanced genomic analysis of the entire spectrum of pediatric brain tumors has heralded an era in which stakeholders in the pediatric neuro-oncology community are being challenged to reconsider their current research and diagnostic and treatment strategies. The incorporation of this new information into the next-generation treatment protocols will unleash new challenges. This review succinctly summarizes the key advances in our understanding of the common pediatric brain tumors (ie, medulloblastoma, low- and high-grade gliomas, diffuse intrinsic pontine glioma, and ependymoma) and some selected rare tumors (ie, atypical teratoid/rhabdoid tumor and CNS primitive neuroectodermal tumor). The potential impact of this new information on future clinical protocols also is discussed. Cutting-edge genomics technologies and the information gained from such studies are facilitating the identification of molecularly defined subgroups within patients with particular pediatric brain tumors. The number of evaluable patients in each subgroup is small, particularly in the subgroups of rare diseases. Therefore, international collaboration will be crucial to draw meaningful conclusions about novel approaches to treating pediatric brain tumors.
© 2015 by American Society of Clinical Oncology.
Conflict of interest statement
Authors' disclosures of potential conflicts of interest are found in the article online at
Figures
References
-
- Gajjar AJ, Robinson GW. Medulloblastoma: Translating discoveries from the bench to the bedside. Nat Rev Clin Oncol. 2014;11:714–722. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
