

Fluorescence Turn-On Folding Sensor To Monitor Proteome Stress in Live Cells
- PMID: 26305239
- PMCID: PMC4755273
- DOI: 10.1021/jacs.5b04366
Fluorescence Turn-On Folding Sensor To Monitor Proteome Stress in Live Cells
Abstract
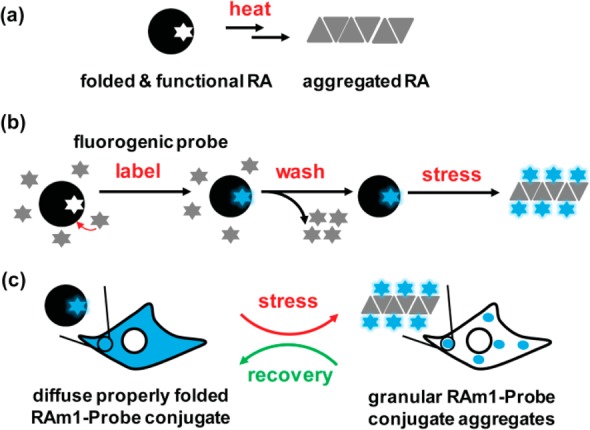
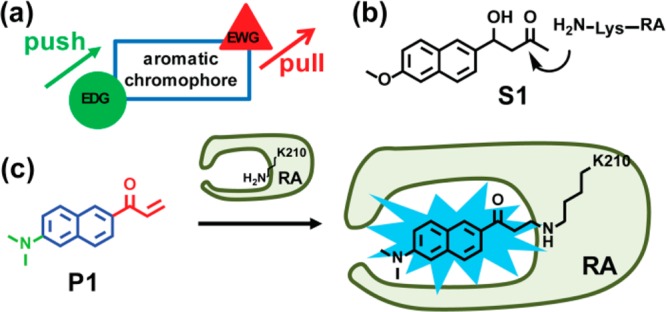
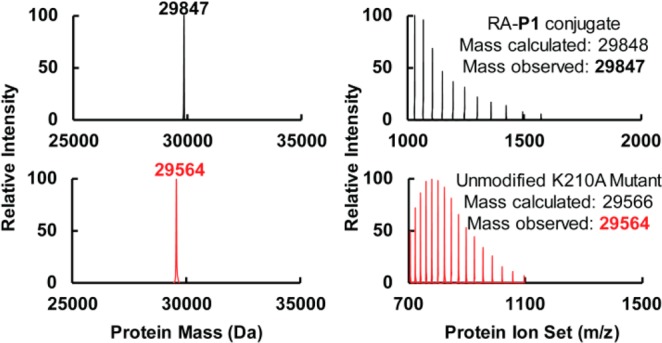
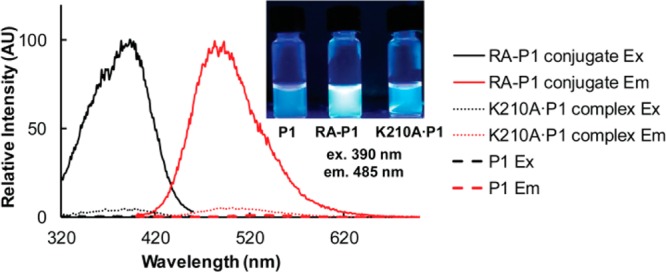
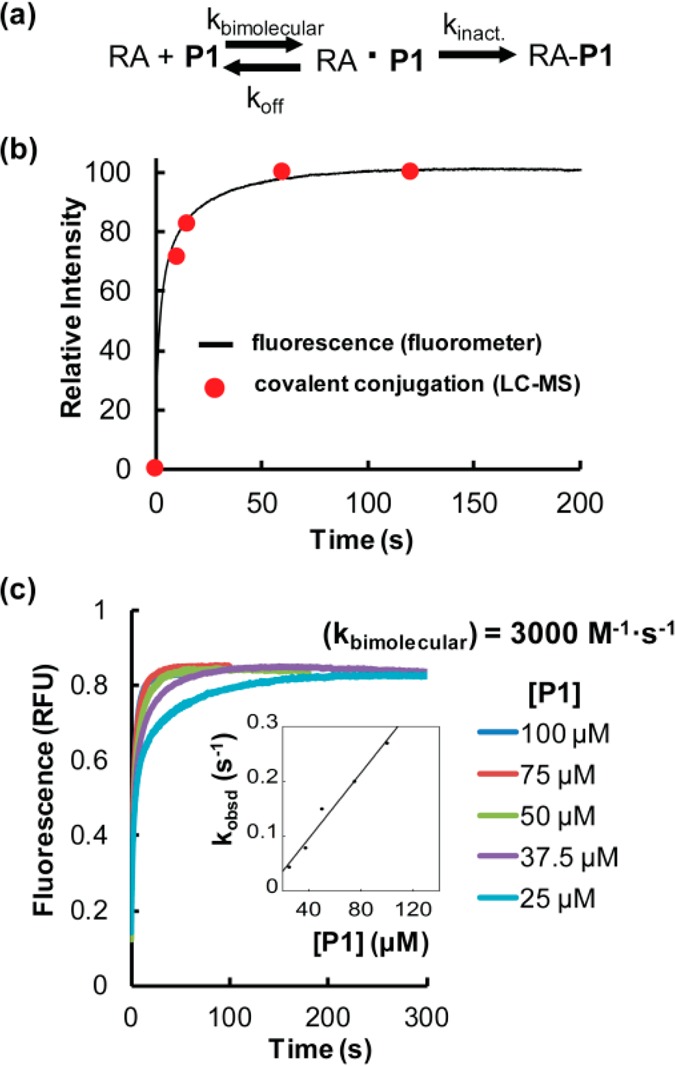
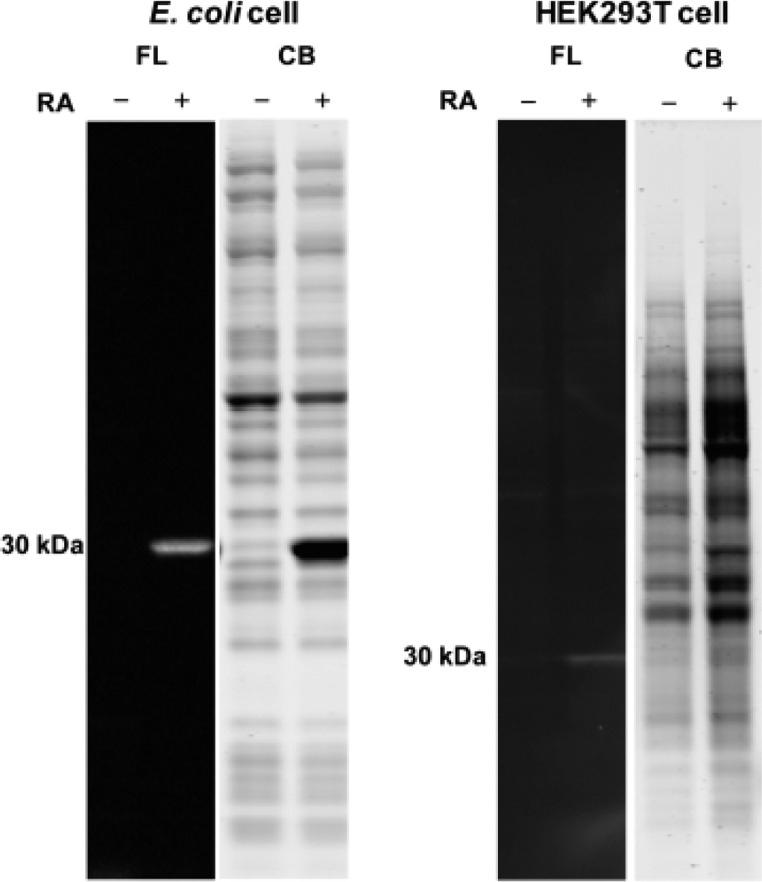
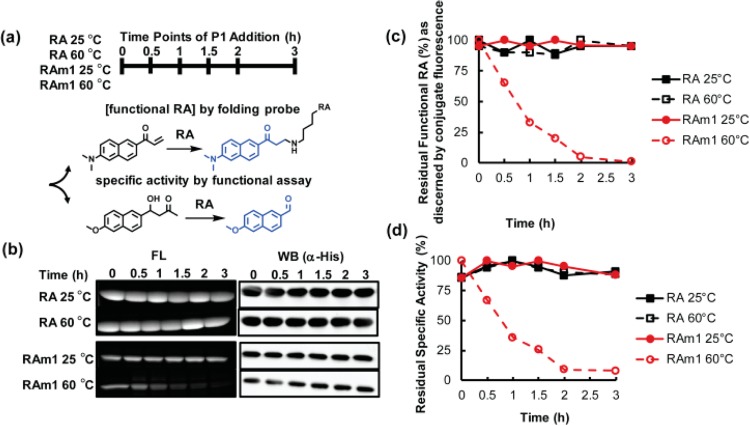
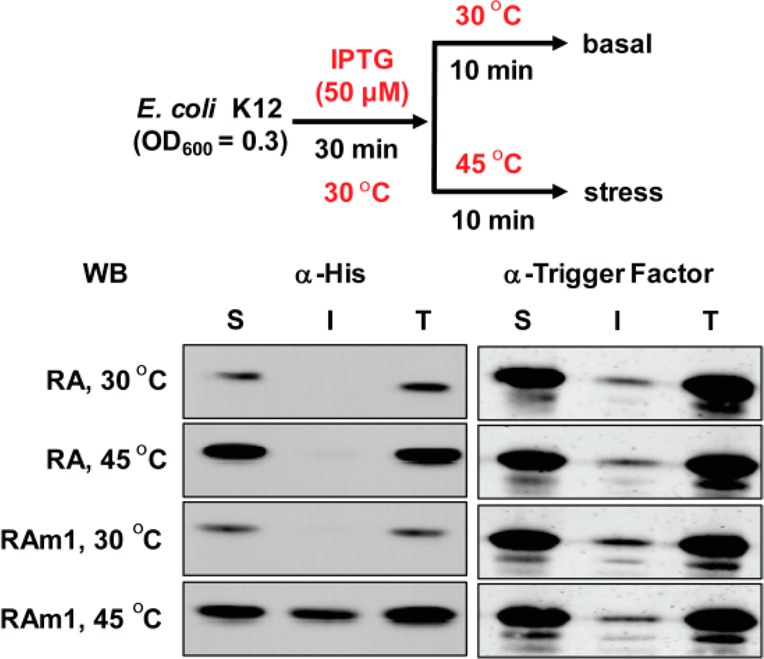
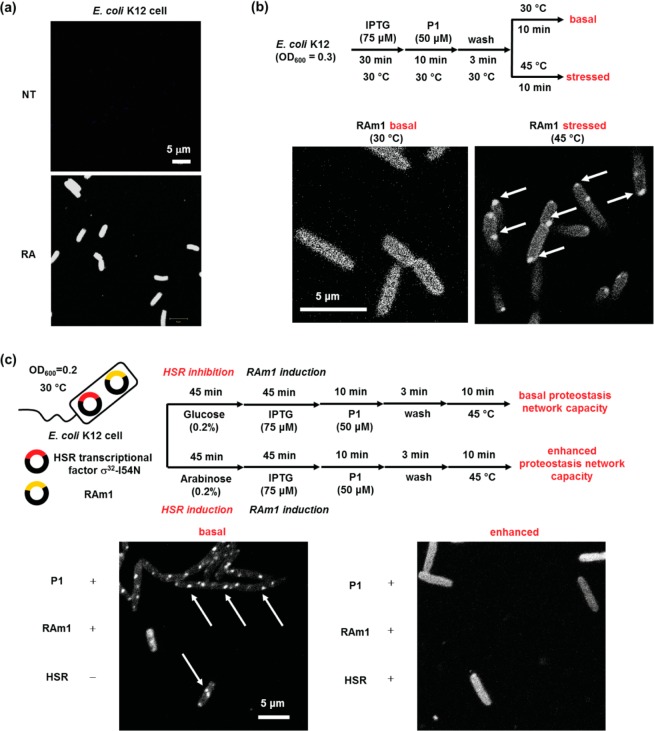
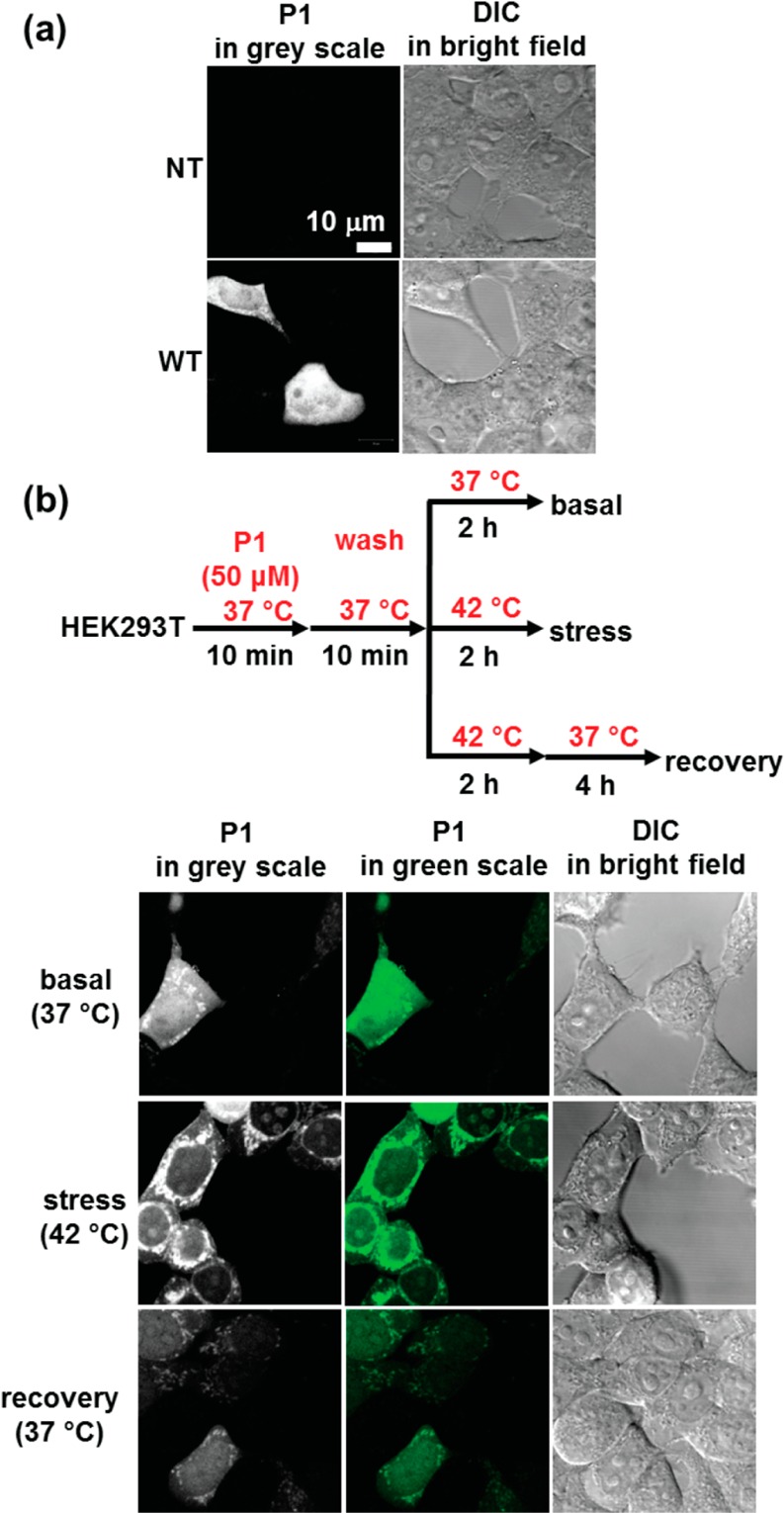
Proteome misfolding and/or aggregation, caused by a thermal perturbation or a related stress, transiently challenges the cellular protein homeostasis (proteostasis) network capacity of cells by consuming chaperone/chaperonin pathway and degradation pathway capacity. Developing protein client-based probes to quantify the cellular proteostasis network capacity in real time is highly desirable. Herein we introduce a small-molecule-regulated fluorescent protein folding sensor based on a thermo-labile mutant of the de novo designed retroaldolase (RA) enzyme. Since RA enzyme activity is not present in any cell, the protein folding sensor is bioorthogonal. The fluorogenic small molecule was designed to become fluorescent when it binds to and covalently reacts with folded and functional RA. Thus, in the first experimental paradigm, cellular proteostasis network capacity and its dynamics are reflected by RA-small molecule conjugate fluorescence, which correlates with the amount of folded and functional RA present, provided that pharmacologic chaperoning is minimized. In the second experimental scenario, the RA-fluorogenic probe conjugate is pre-formed in a cell by simply adding the fluorogenic probe to the cell culture media. Unreacted probe is then washed away before a proteome misfolding stress is applied in a pulse-chase-type experiment. Insufficient proteostasis network capacity is reflected by aggregate formation of the fluorescent RA-fluorogenic probe conjugate. Removal of the stress results in apparent RA-fluorogenic probe conjugate re-folding, mediated in part by the heat-shock response transcriptional program augmenting cytosolic proteostasis network capacity, and in part by time-dependent RA-fluorogenic probe conjugate degradation by cellular proteolysis.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
