

Comparison of Hi-C results using in-solution versus in-nucleus ligation
- PMID: 26306623
- PMCID: PMC4580221
- DOI: 10.1186/s13059-015-0753-7
Comparison of Hi-C results using in-solution versus in-nucleus ligation
Abstract
Background: Chromosome conformation capture and various derivative methods such as 4C, 5C and Hi-C have emerged as standard tools to analyze the three-dimensional organization of the genome in the nucleus. These methods employ ligation of diluted cross-linked chromatin complexes, intended to favor proximity-dependent, intra-complex ligation. During development of single-cell Hi-C, we devised an alternative Hi-C protocol with ligation in preserved nuclei rather than in solution. Here we directly compare Hi-C methods employing in-nucleus ligation with the standard in-solution ligation.
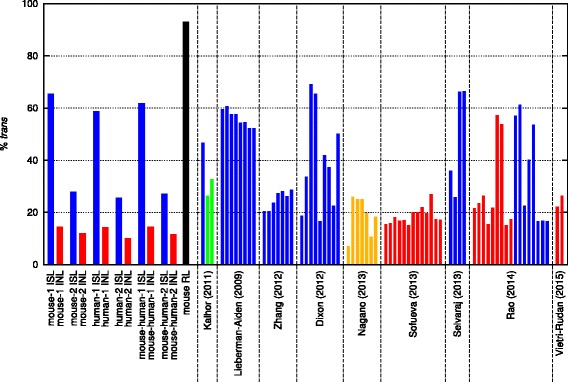
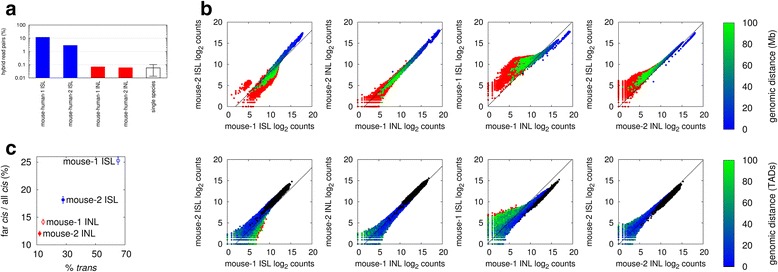
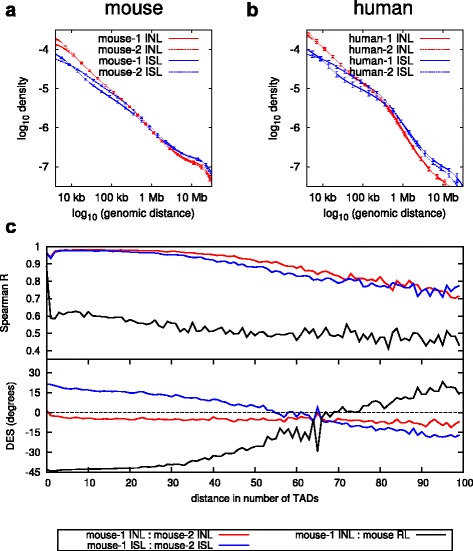
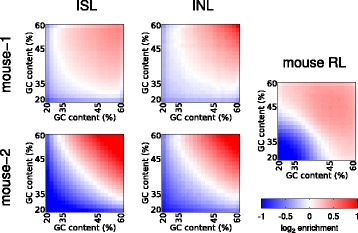
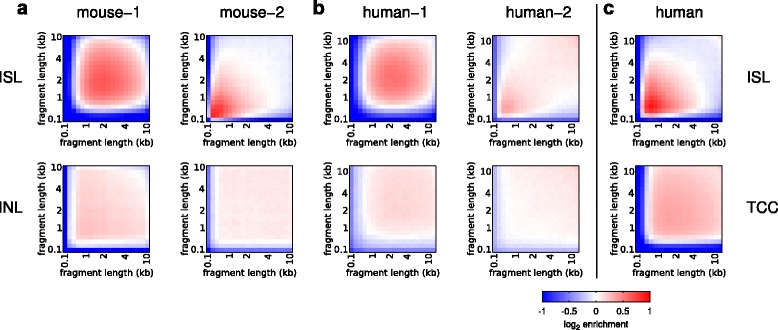
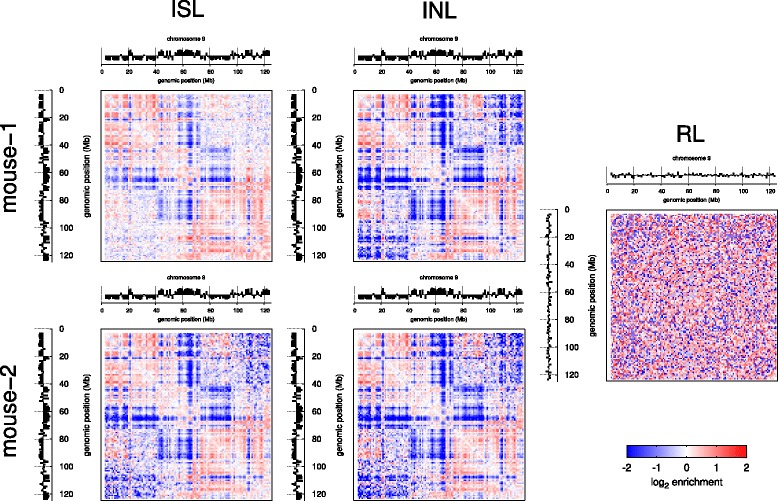
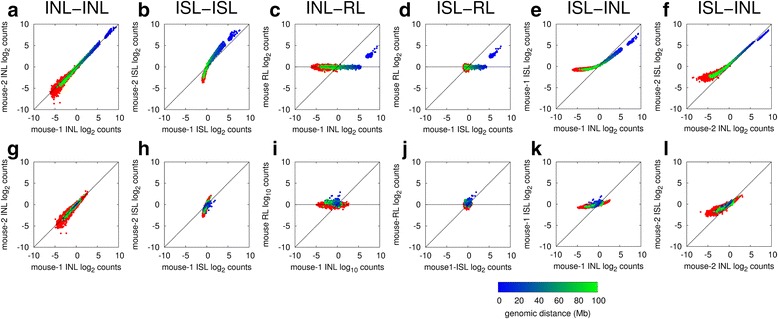
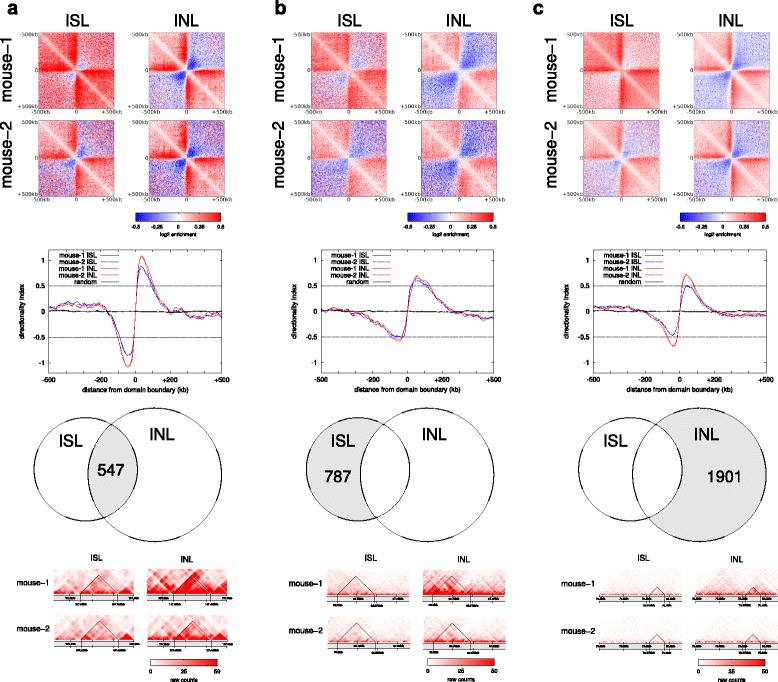
Results: We show in-nucleus ligation results in consistently lower levels of inter-chromosomal contacts. Through chromatin mixing experiments we show that a significantly large fraction of inter-chromosomal contacts are the result of spurious ligation events formed during in-solution ligation. In-nucleus ligation significantly reduces this source of experimental noise, and results in improved reproducibility between replicates. We also find that in-nucleus ligation eliminates restriction fragment length bias found with in-solution ligation. These improvements result in greater reproducibility of long-range intra-chromosomal and inter-chromosomal contacts, as well as enhanced detection of structural features such as topologically associated domain boundaries.
Conclusions: We conclude that in-nucleus ligation captures chromatin interactions more consistently over a wider range of distances, and significantly reduces both experimental noise and bias. In-nucleus ligation creates higher quality Hi-C libraries while simplifying the experimental procedure. We suggest that the entire range of 3C applications are likely to show similar benefits from in-nucleus ligation.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
