

Whole Exome Sequencing Reveals Mutations in Known Retinal Disease Genes in 33 out of 68 Israeli Families with Inherited Retinopathies
- PMID: 26306921
- PMCID: PMC4549705
- DOI: 10.1038/srep13187
Whole Exome Sequencing Reveals Mutations in Known Retinal Disease Genes in 33 out of 68 Israeli Families with Inherited Retinopathies
Abstract
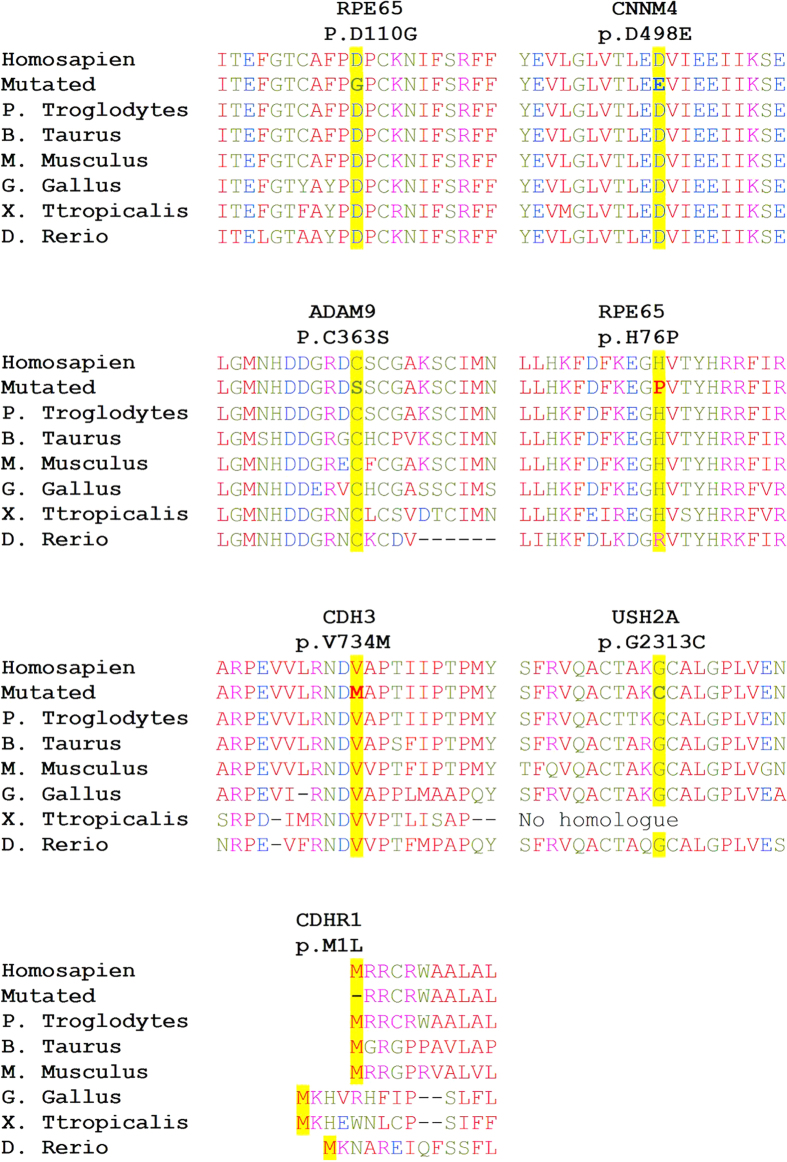
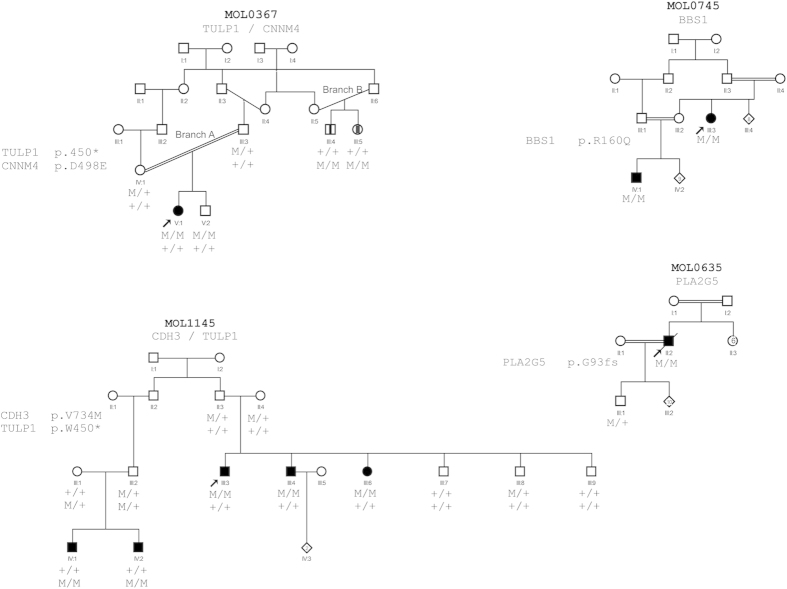
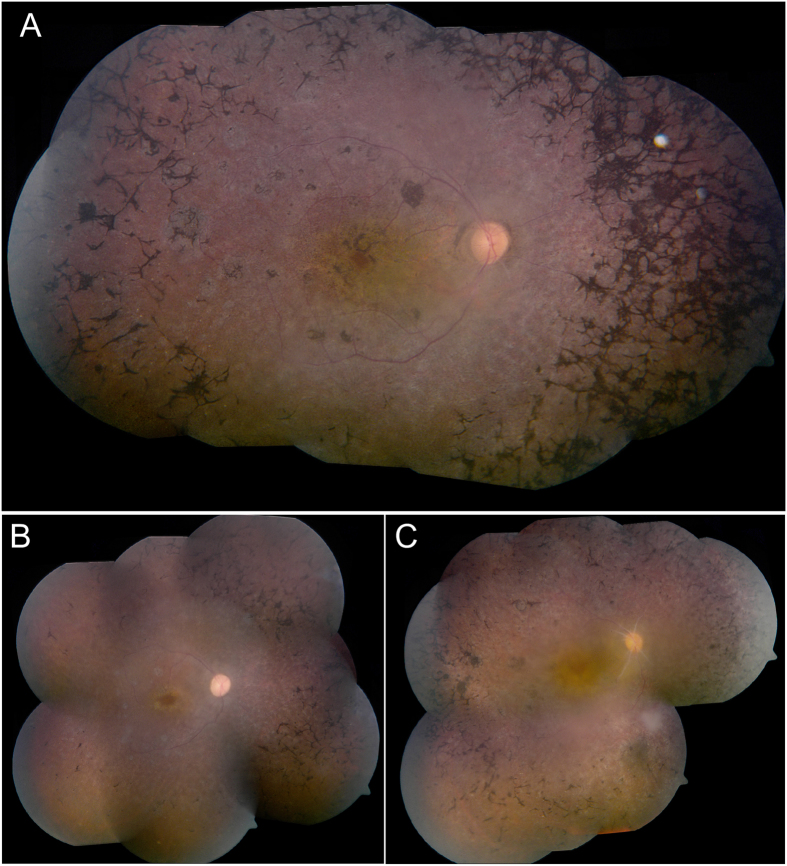
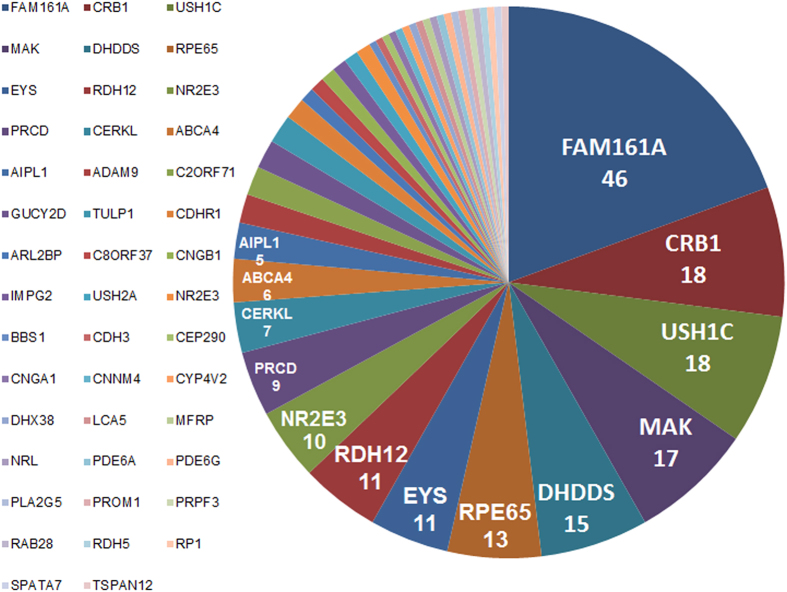
Whole exome sequencing (WES) is a powerful technique for identifying sequence changes in the human genome. The goal of this study was to delineate the genetic defects in patients with inherited retinal diseases (IRDs) using WES. WES was performed on 90 patient DNA samples from 68 families and 226 known genes for IRDs were analyzed. Sanger sequencing was used to validate potential pathogenic variants that were also subjected to segregation analysis in families. Thirty-three causative mutations (19 novel and 14 known) in 25 genes were identified in 33 of the 68 families. The vast majority of mutations (30 out of 33) have not been reported in the Israeli and the Palestinian populations. Nine out of the 33 mutations were detected in additional families from the same ethnic population, suggesting a founder effect. In two families, identified phenotypes were different from the previously reported clinical findings associated with the causative gene. This is the largest genetic analysis of IRDs in the Israeli and Palestinian populations to date. We also demonstrate that WES is a powerful tool for rapid analysis of known disease genes in large patient cohorts.
Figures
References
-
- Simonelli F. et al. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest ophthalmol Vis Sci 48, 4284–4290 (2007). - PubMed
-
- den Hollander A. I., Roepman R., Koenekoop R. K. & Cremers F. P. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 27, 391–419 (2008). - PubMed
-
- Rosenberg T. Epidemiology of hereditary ocular disorders. Dev Ophthalmol 37, 16–33 (2003). - PubMed
-
- Peterlin B. et al. Prevalence of retinitis pigmentosa in Slovenia. Clin Genet 42, 122–123 (1992). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
