

Chlamydomonas Genome Resource for Laboratory Strains Reveals a Mosaic of Sequence Variation, Identifies True Strain Histories, and Enables Strain-Specific Studies
- PMID: 26307380
- PMCID: PMC4815092
- DOI: 10.1105/tpc.15.00508
Chlamydomonas Genome Resource for Laboratory Strains Reveals a Mosaic of Sequence Variation, Identifies True Strain Histories, and Enables Strain-Specific Studies
Abstract
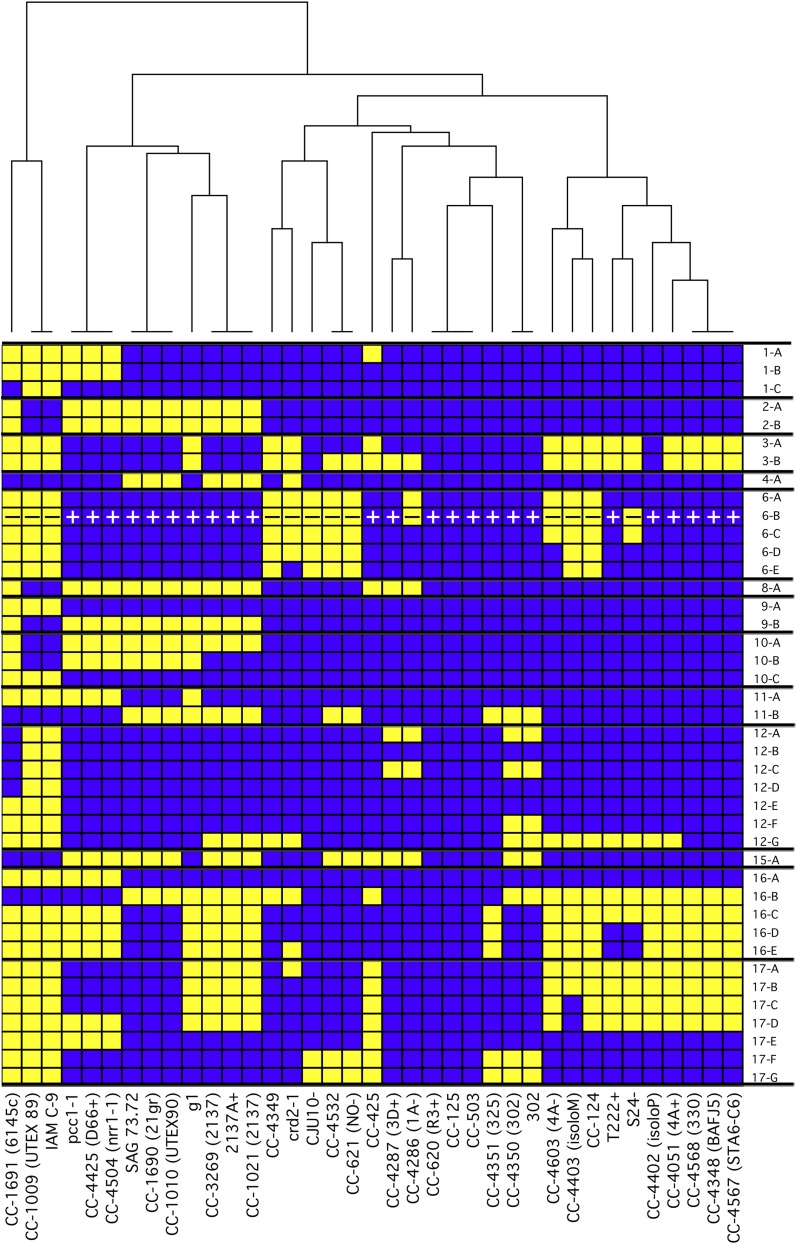
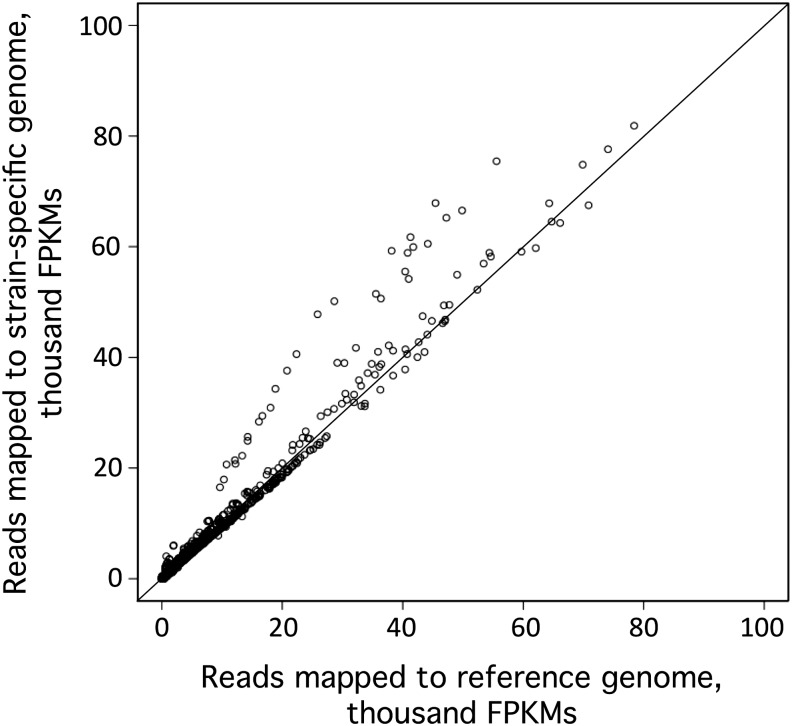
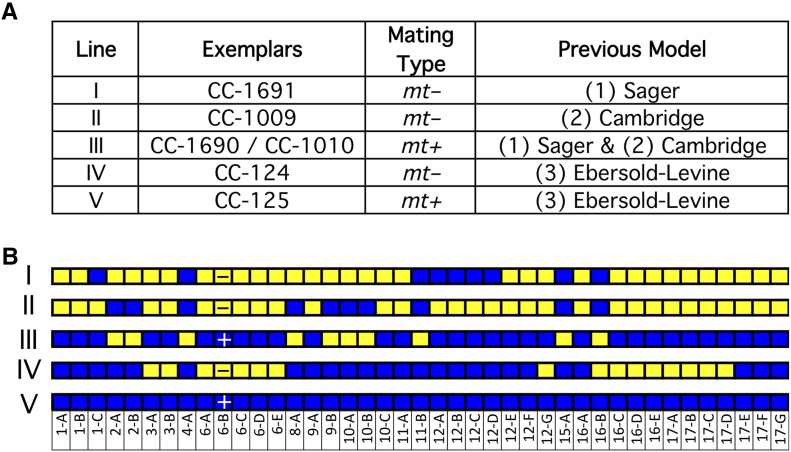
Chlamydomonas reinhardtii is a widely used reference organism in studies of photosynthesis, cilia, and biofuels. Most research in this field uses a few dozen standard laboratory strains that are reported to share a common ancestry, but exhibit substantial phenotypic differences. In order to facilitate ongoing Chlamydomonas research and explain the phenotypic variation, we mapped the genetic diversity within these strains using whole-genome resequencing. We identified 524,640 single nucleotide variants and 4812 structural variants among 39 commonly used laboratory strains. Nearly all (98.2%) of the total observed genetic diversity was attributable to the presence of two, previously unrecognized, alternate haplotypes that are distributed in a mosaic pattern among the extant laboratory strains. We propose that these two haplotypes are the remnants of an ancestral cross between two strains with ∼2% relative divergence. These haplotype patterns create a fingerprint for each strain that facilitates the positive identification of that strain and reveals its relatedness to other strains. The presence of these alternate haplotype regions affects phenotype scoring and gene expression measurements. Here, we present a rich set of genetic differences as a community resource to allow researchers to more accurately conduct and interpret their experiments with Chlamydomonas.
© 2015 American Society of Plant Biologists. All rights reserved.
Figures
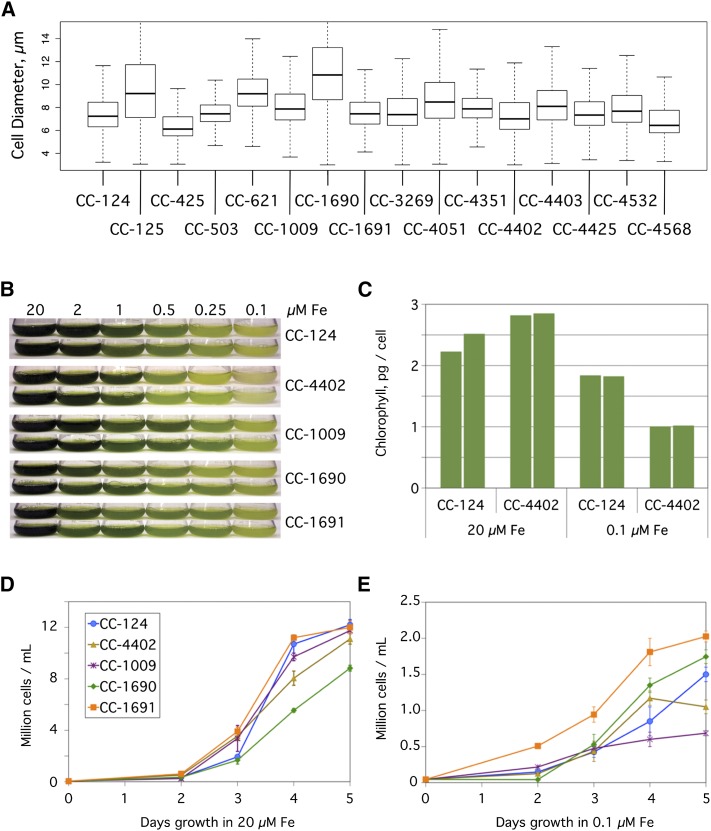
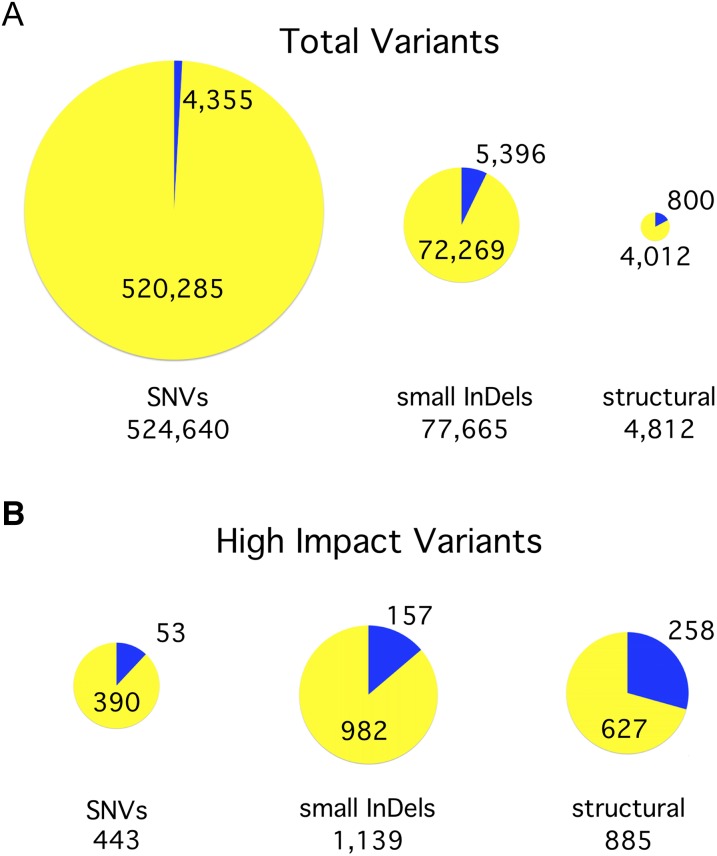
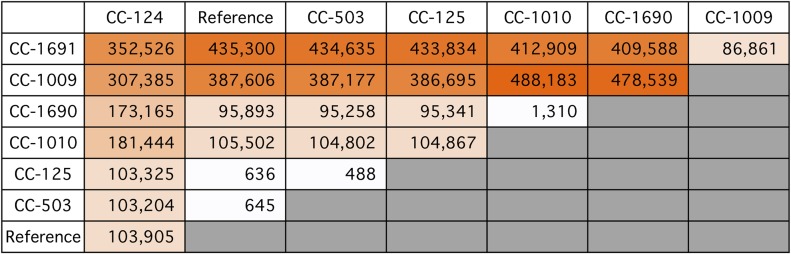
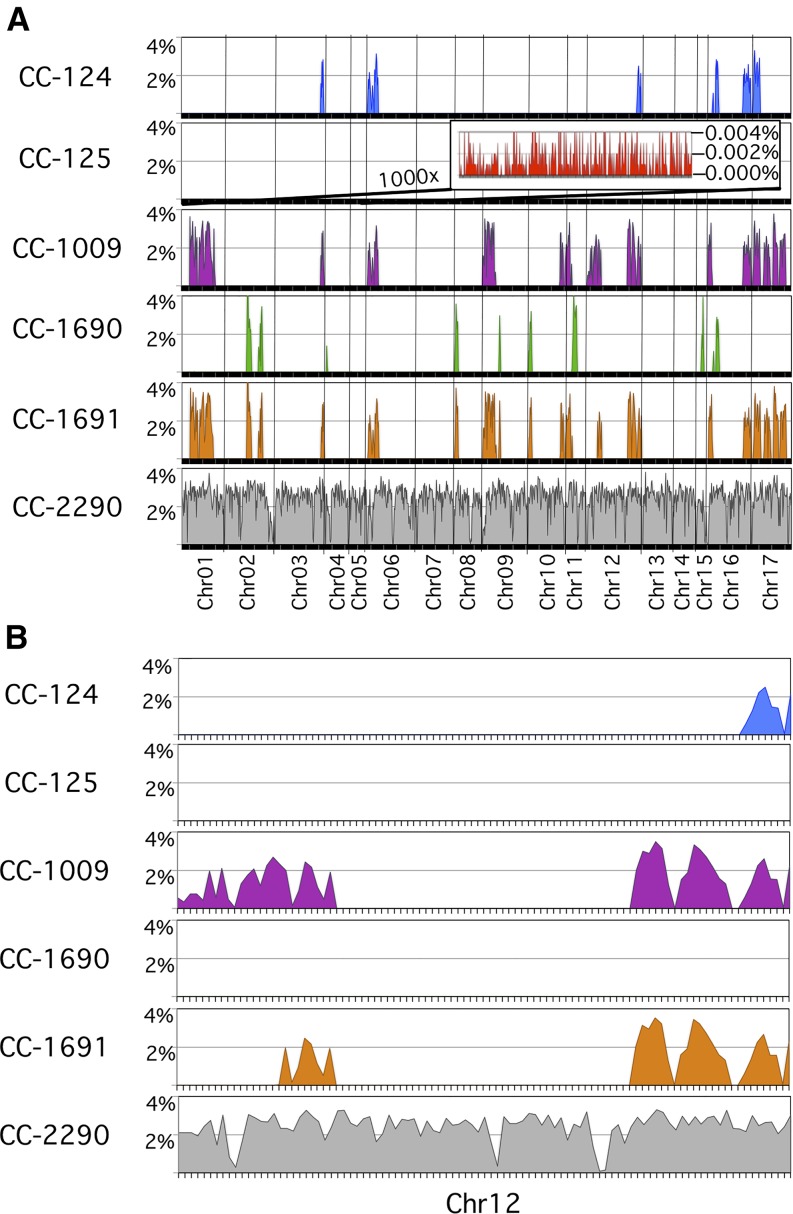

Comment in
-
Genomic Diversity in Chlamydomonas Laboratory and Field Strains.Plant Cell. 2015 Sep;27(9):2315-6. doi: 10.1105/tpc.15.00778. Epub 2015 Sep 21. Plant Cell. 2015. PMID: 26392079 Free PMC article. No abstract available.
References
-
- Cakmak T., Angun P., Demiray Y.E., Ozkan A.D., Elibol Z., Tekinay T. (2012). Differential effects of nitrogen and sulfur deprivation on growth and biodiesel feedstock production of Chlamydomonas reinhardtii. Biotechnol. Bioeng. 109: 1947–1957. - PubMed
-
- Castruita M., Casero D., Karpowicz S.J., Kropat J., Vieler A., Hsieh S.I., Yan W., Cokus S., Loo J.A., Benning C., Pellegrini M., Merchant S.S. (2011). Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 23: 1273–1292. - PMC - PubMed
-
- Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6: 80–92. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- SRA/SRP053354
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
