

Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene
- PMID: 26310628
- PMCID: PMC4643641
- DOI: 10.1093/brain/awv241
Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene
Abstract
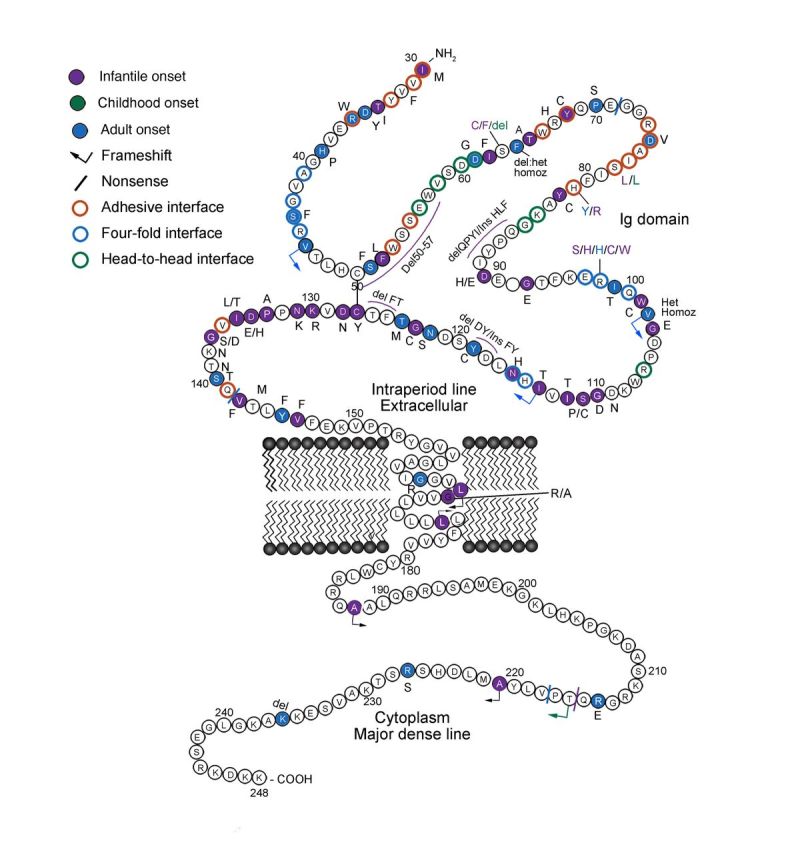
We aimed to characterize genotype-phenotype correlations and establish baseline clinical data for peripheral neuropathies caused by mutations in the myelin protein zero (MPZ) gene. MPZ mutations are the second leading cause of Charcot-Marie-Tooth disease type 1. Recent research makes clinical trials for patients with MPZ mutations a realistic possibility. However, the clinical severity varies with different mutations and natural history data on progression is sparse. We present cross-sectional data to begin to define the phenotypic spectrum and clinical baseline of patients with these mutations. A cohort of patients with MPZ gene mutations was identified in 13 centres of the Inherited Neuropathies Consortium - Rare Disease Clinical Research Consortium (INC-RDCRC) between 2009 and 2012 and at Wayne State University between 1996 and 2009. Patient phenotypes were quantified by the Charcot-Marie-Tooth disease neuropathy score version 1 or 2 and the Charcot-Marie-Tooth disease paediatric scale outcome instruments. Genetic testing was performed in all patients and/or in first- or second-degree relatives to document mutation in MPZ gene indicating diagnosis of Charcot-Marie-Tooth disease type 1B. There were 103 patients from 71 families with 47 different MPZ mutations with a mean age of 40 years (range 3-84 years). Patients and mutations were separated into infantile, childhood and adult-onset groups. The infantile onset group had higher Charcot-Marie-Tooth disease neuropathy score version 1 or 2 and slower nerve conductions than the other groups, and severity increased with age. Twenty-three patients had no family history of Charcot-Marie-Tooth disease. Sixty-one patients wore foot/ankle orthoses, 19 required walking assistance or support, and 10 required wheelchairs. There was hearing loss in 21 and scoliosis in 17. Forty-two patients did not begin walking until after 15 months of age. Half of the infantile onset patients then required ambulation aids or wheelchairs for ambulation. Our results demonstrate that virtually all MPZ mutations are associated with specific phenotypes. Early onset (infantile and childhood) phenotypes likely represent developmentally impaired myelination, whereas the adult-onset phenotype reflects axonal degeneration without antecedent demyelination. Data from this cohort of patients will provide the baseline data necessary for clinical trials of patients with Charcot-Marie-Tooth disease caused by MPZ gene mutations.
Keywords: CMT1B; MPZ; demyelination; myelin, neuropathy.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures



References
-
- Bai Y, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, et al. Effect of an R69C mutation in the myelin protein zero gene on myelination and ion channel subtypes. Arch Neurol 2006; 63: 1787–94. - PubMed
-
- Baloh RH, Jen JC, Kim G, Baloh RW. Chronic cough due to Thr124Met mutation in the peripheral myelin protein zero (MPZ gene). Neurology 2004; 62: 1905–6. - PubMed
-
- Becker PE, editor. Humangenetik. Ein kurzes Handbuch; Stuttgard: Thieme; 1978. p. 425.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
