

A Coalescent Model for a Sweep of a Unique Standing Variant
- PMID: 26311475
- PMCID: PMC4596678
- DOI: 10.1534/genetics.115.178962
A Coalescent Model for a Sweep of a Unique Standing Variant
Abstract
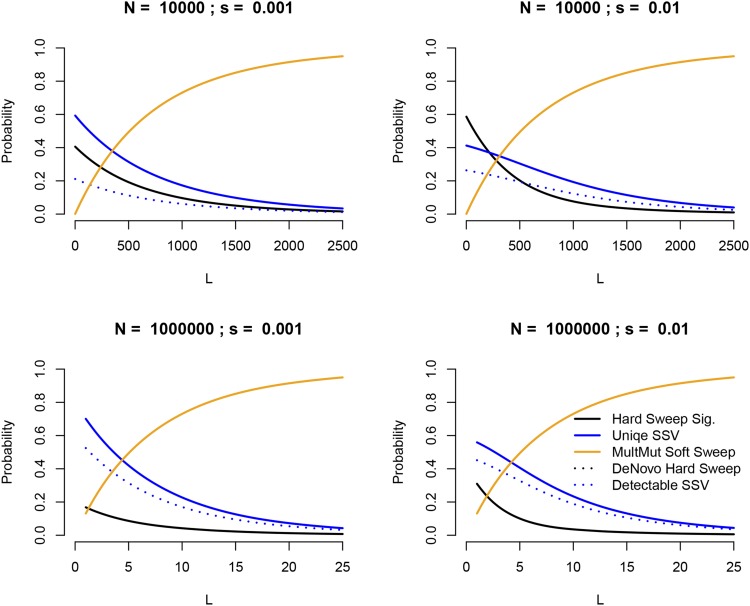
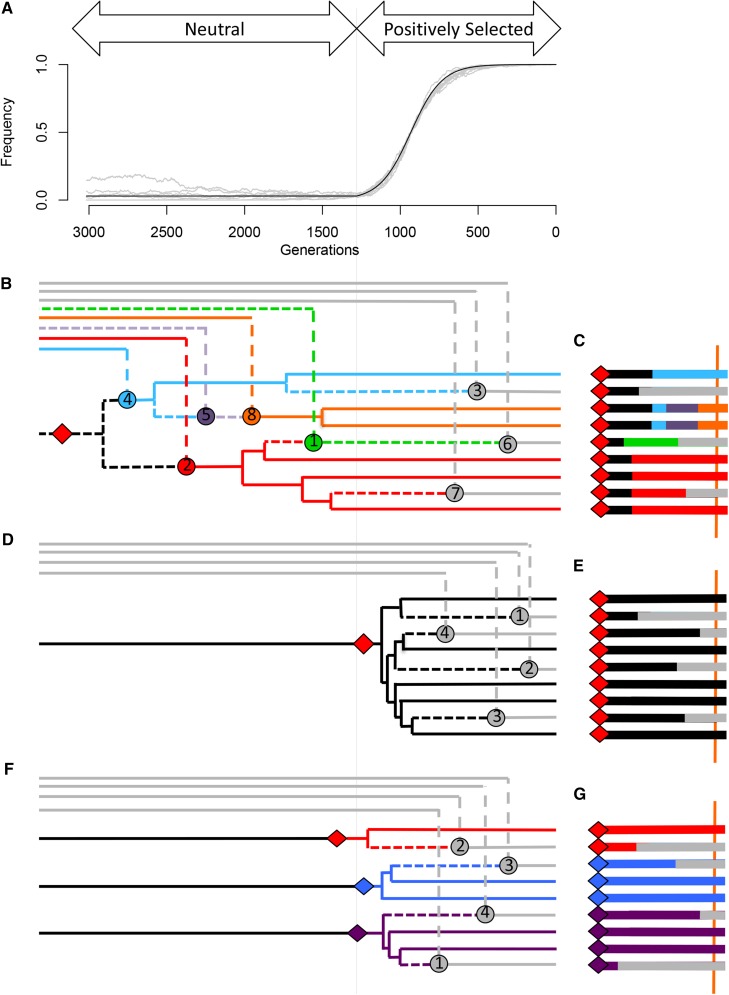
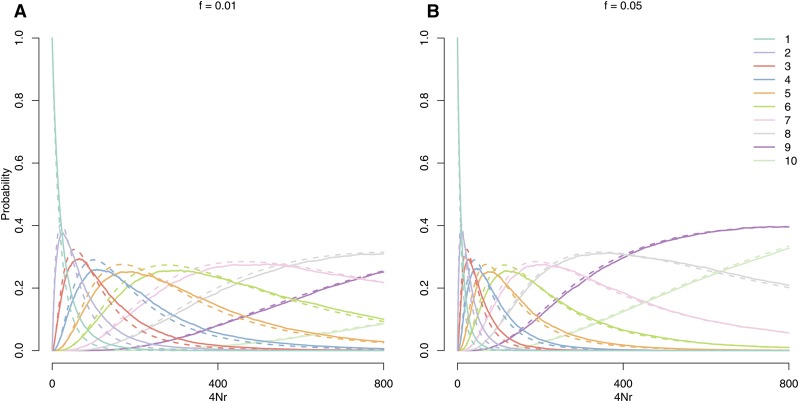
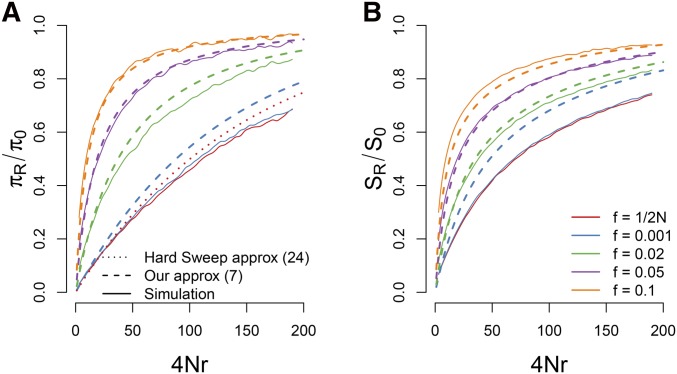
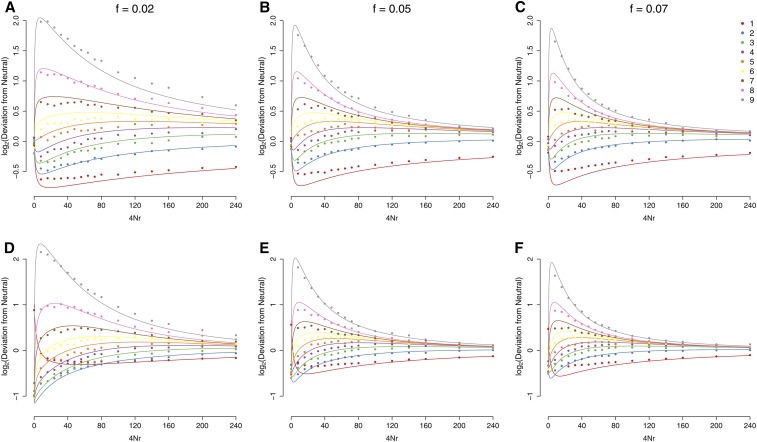
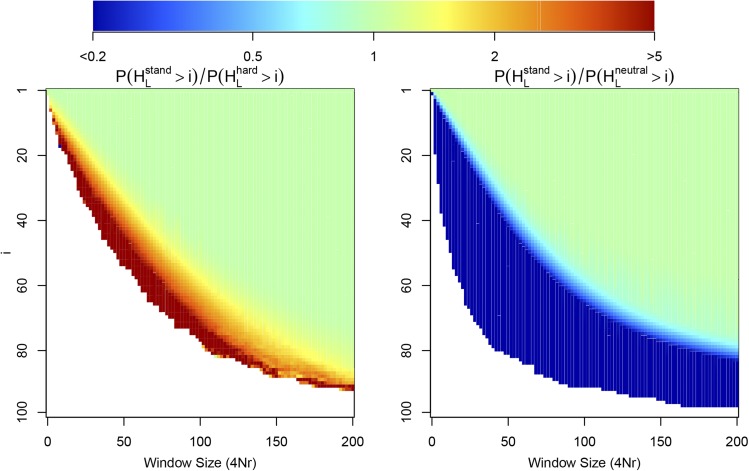
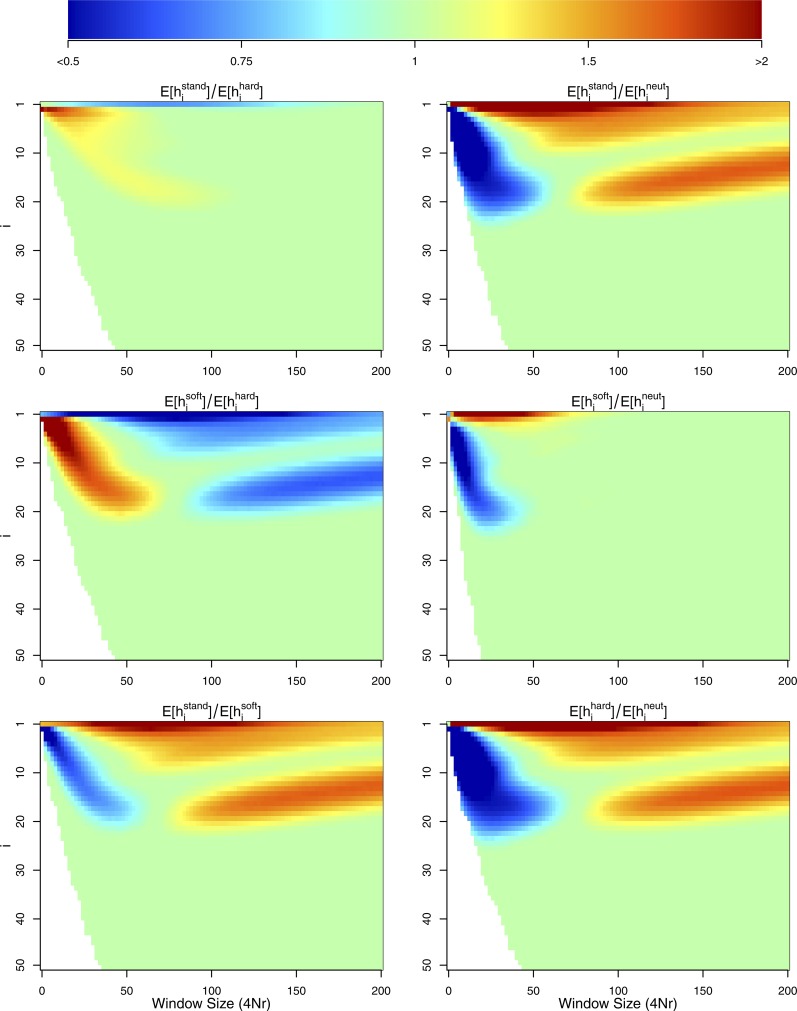
The use of genetic polymorphism data to understand the dynamics of adaptation and identify the loci that are involved has become a major pursuit of modern evolutionary genetics. In addition to the classical "hard sweep" hitchhiking model, recent research has drawn attention to the fact that the dynamics of adaptation can play out in a variety of different ways and that the specific signatures left behind in population genetic data may depend somewhat strongly on these dynamics. One particular model for which a large number of empirical examples are already known is that in which a single derived mutation arises and drifts to some low frequency before an environmental change causes the allele to become beneficial and sweeps to fixation. Here, we pursue an analytical investigation of this model, bolstered and extended via simulation study. We use coalescent theory to develop an analytical approximation for the effect of a sweep from standing variation on the genealogy at the locus of the selected allele and sites tightly linked to it. We show that the distribution of haplotypes that the selected allele is present on at the time of the environmental change can be approximated by considering recombinant haplotypes as alleles in the infinite-alleles model. We show that this approximation can be leveraged to make accurate predictions regarding patterns of genetic polymorphism following such a sweep. We then use simulations to highlight which sources of haplotypic information are likely to be most useful in distinguishing this model from neutrality, as well as from other sweep models, such as the classic hard sweep and multiple-mutation soft sweeps. We find that in general, adaptation from a unique standing variant will likely be difficult to detect on the basis of genetic polymorphism data from a single population time point alone, and when it can be detected, it will be difficult to distinguish from other varieties of selective sweeps. Samples from multiple populations and/or time points have the potential to ease this difficulty.
Keywords: coalescent theory; genetic hitchhiking; natural selection; soft sweep; standing variation.
Copyright © 2015 by the Genetics Society of America.
Figures
References
-
- Bank C., Ewing G. B., Ferrer-Admettla A., Foll M., Jensen J. D., 2014. Thinking too positive? Revisiting current methods of population genetic selection inference. Trends Genet. 30(12): 540–546. - PubMed
-
- Barrett R. D. H., Schluter D., 2008. Adaptation from standing genetic variation. Trends Ecol. Evol. 23(1): 38–44. - PubMed
-
- Barton N. H., 1998. The effect of hitch-hiking on neutral genealogies. Genet. Res. 72: 123–133.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
