

Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants
- PMID: 26312827
- PMCID: PMC4857209
- DOI: 10.1038/gim.2015.108
Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants
Abstract
Purpose: The purpose of this study was to enhance understanding of lysosomal acid lipase deficiency (LALD) in infancy.
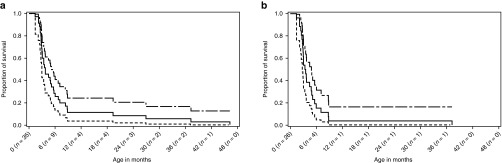
Methods: Investigators reviewed medical records of infants with LALD and summarized data for the overall population and for patients with and without early growth failure (GF). Kaplan-Meier survival analyses were conducted for the overall population and for treated and untreated patients.
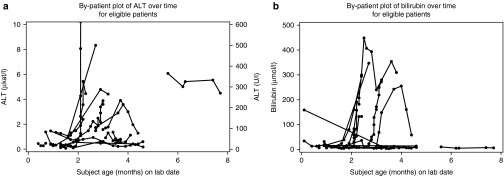
Results: Records for 35 patients, 26 with early GF, were analyzed. Prominent symptom manifestations included vomiting, diarrhea, and steatorrhea. Median age at death was 3.7 months; estimated probability of survival past age 12 months was 0.114 (95% confidence interval (CI): 0.009-0.220). Among patients with early GF, median age at death was 3.5 months; estimated probability of survival past age 12 months was 0.038 (95% CI: 0.000-0.112). Treated patients (hematopoietic stem cell transplant (HSCT), n = 9; HSCT and liver transplant, n = 1) in the overall population and the early GF subset survived longer than untreated patients, but survival was still poor (median age at death, 8.6 months).
Conclusions: These data confirm and expand earlier insights on the progression and course of LALD presenting in infancy. Despite variations in the nature, onset, and severity of clinical manifestations, and treatment attempts, clinical outcome was poor.Genet Med 18 5, 452-458.
Figures
References
-
- Lysosomal acid lipase deficiency (#278000). http://www.omim.org/entry/278000. Published 18 July 2012. Accessed 19 November 2013.
-
- Grabowski G, Charnas L, Du H. Lysosomal acid lipase deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum. In: Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A (eds). The Metabolic and Molecular Basis of Inherited Metabolic Disease, 8th edn. McGraw-Hill: New York, 2012. http://ommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374143. Accessed 21 November 2013.
-
- Wolman M. Wolman disease and its treatment. Clin Pediatr (Phila) 1995;34:207–212. - PubMed
-
- Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013;58:1230–1243. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
