

Nonlinearities in protein space limit the utility of informatics in protein biophysics
- PMID: 26315852
- PMCID: PMC4609284
- DOI: 10.1002/prot.24916
Nonlinearities in protein space limit the utility of informatics in protein biophysics
Abstract
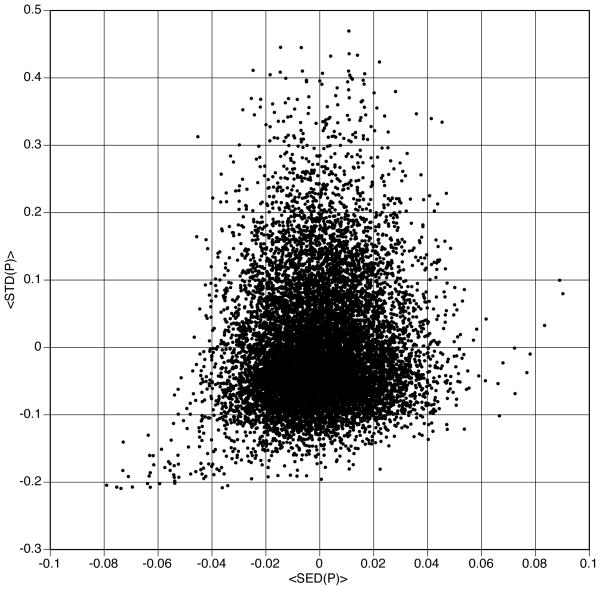
We examine the utility of informatic-based methods in computational protein biophysics. To do so, we use newly developed metric functions to define completely independent sequence and structure spaces for a large database of proteins. By investigating the relationship between these spaces, we demonstrate quantitatively the limits of knowledge-based correlation between the sequences and structures of proteins. It is shown that there are well-defined, nonlinear regions of protein space in which dissimilar structures map onto similar sequences (the conformational switch), and dissimilar sequences map onto similar structures (remote homology). These nonlinearities are shown to be quite common-almost half the proteins in our database fall into one or the other of these two regions. They are not anomalies, but rather intrinsic properties of structural encoding in amino acid sequences. It follows that extreme care must be exercised in using bioinformatic data as a basis for computational structure prediction. The implications of these results for protein evolution are examined.
Keywords: Fourier analysis; conformational switches; distant homology; sequence space; structure space.
© 2015 Wiley Periodicals, Inc.
Figures
References
-
- Yang Y, Faraggi E, Zhao H, Zhou Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076–2082. - PMC - PubMed
-
- Ben-Hur A, Brutlag D. Remote homology detection: a motif based approach. Bioinformatics. 2003;19(Suppl 1):i26–i33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
