

The Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia
- PMID: 26316429
- PMCID: PMC4563192
- DOI: 10.4274/jcrpe.1891
The Diagnosis and Management of Hyperinsulinaemic Hypoglycaemia
Abstract
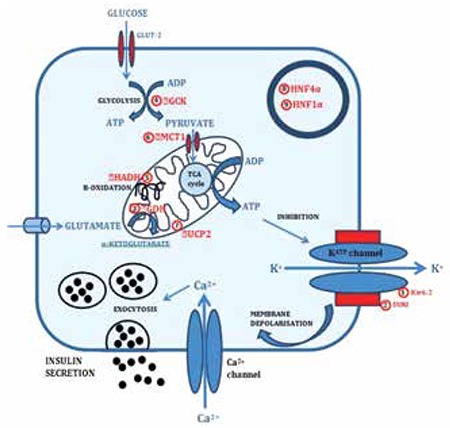
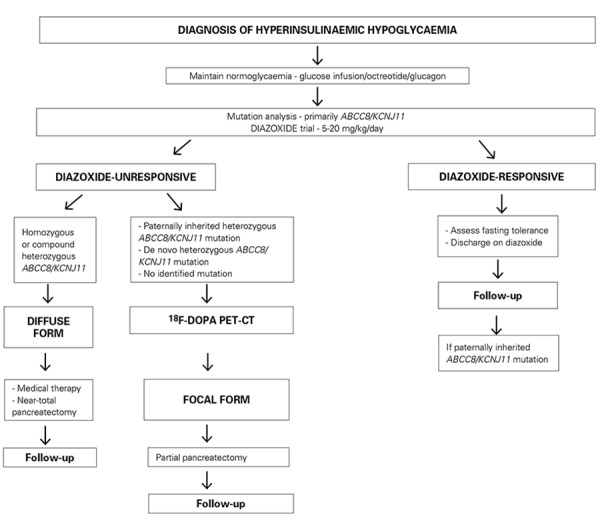
Insulin secretion from pancreatic β-cells is tightly regulated to keep fasting blood glucose concentrations within the normal range (3.5-5.5 mmol/L). Hyperinsulinaemic hypoglycaemia (HH) is a heterozygous condition in which insulin secretion becomes unregulated and its production persists despite low blood glucose levels. It is the most common cause of severe and persistent hypoglycaemia in neonates and children. The most severe and permanent forms are due to congenital hyperinsulinism (CHI). Recent advances in genetics have linked CHI to mutations in 9 genes that play a key role in regulating insulin secretion (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, UCP2, HNF4A and HNF1A). Histologically, CHI can be divided into 3 types; diffuse, focal and atypical. Given the biochemical nature of HH (non-ketotic), a delay in the diagnosis and management can result in irreversible brain damage. Therefore, it is essential to diagnose and treat HH promptly. Advances in molecular genetics, imaging methods (18F-DOPA PET-CT), medical therapy and surgical approach (laparoscopic surgery) have completely changed the management and improved the outcome of these children. This review provides an overview of the genetic and molecular mechanisms leading to development of HH in children. The article summarizes the current diagnostic methods and management strategies for the different types of CHI.
Figures
References
-
- Hussain K, Aynsley-Green A. Hyperinsulinism in infancy: understanding the pathophysiology. Int J Biochem Cell Biol. 2003;35:1312–1317. - PubMed
-
- Harris DL, Weston PJ, Harding JE. Lactate, rather than ketones, may provide alternative cerebral fuel in hypoglycaemic newborns. Arch Dis Child Fetal Neonatal Ed. 2015;100:161–164. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous