

Discovery of Drug Synergies in Gastric Cancer Cells Predicted by Logical Modeling
- PMID: 26317215
- PMCID: PMC4567168
- DOI: 10.1371/journal.pcbi.1004426
Discovery of Drug Synergies in Gastric Cancer Cells Predicted by Logical Modeling
Abstract
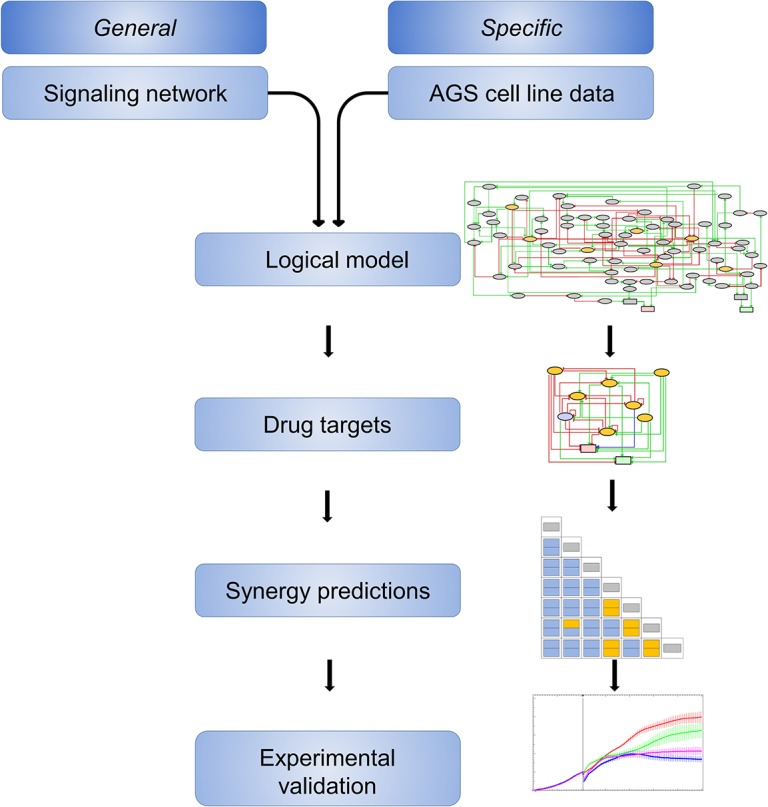
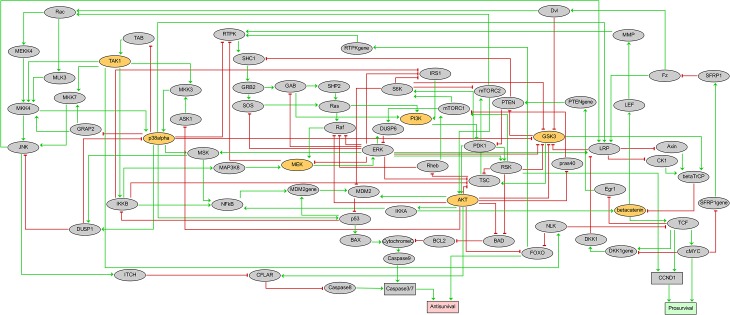
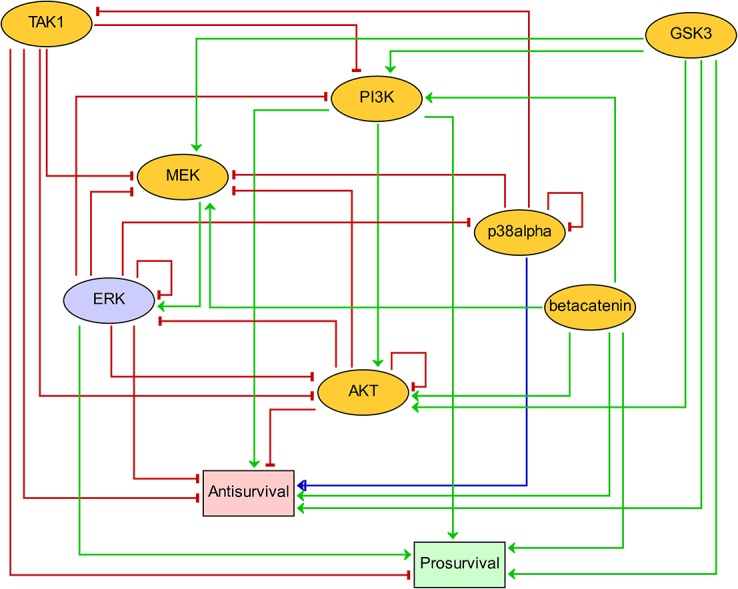
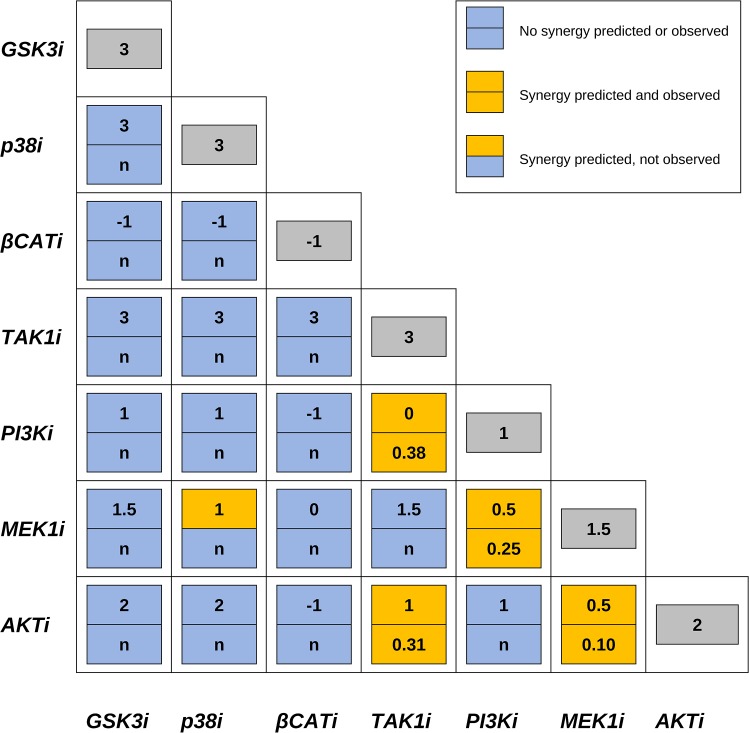
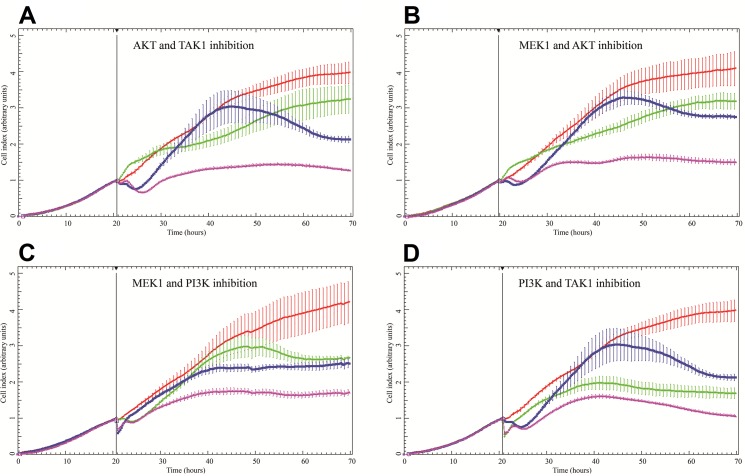
Discovery of efficient anti-cancer drug combinations is a major challenge, since experimental testing of all possible combinations is clearly impossible. Recent efforts to computationally predict drug combination responses retain this experimental search space, as model definitions typically rely on extensive drug perturbation data. We developed a dynamical model representing a cell fate decision network in the AGS gastric cancer cell line, relying on background knowledge extracted from literature and databases. We defined a set of logical equations recapitulating AGS data observed in cells in their baseline proliferative state. Using the modeling software GINsim, model reduction and simulation compression techniques were applied to cope with the vast state space of large logical models and enable simulations of pairwise applications of specific signaling inhibitory chemical substances. Our simulations predicted synergistic growth inhibitory action of five combinations from a total of 21 possible pairs. Four of the predicted synergies were confirmed in AGS cell growth real-time assays, including known effects of combined MEK-AKT or MEK-PI3K inhibitions, along with novel synergistic effects of combined TAK1-AKT or TAK1-PI3K inhibitions. Our strategy reduces the dependence on a priori drug perturbation experimentation for well-characterized signaling networks, by demonstrating that a model predictive of combinatorial drug effects can be inferred from background knowledge on unperturbed and proliferating cancer cells. Our modeling approach can thus contribute to preclinical discovery of efficient anticancer drug combinations, and thereby to development of strategies to tailor treatment to individual cancer patients.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
