

Viral and Cellular Genomes Activate Distinct DNA Damage Responses
- PMID: 26317467
- PMCID: PMC4681434
- DOI: 10.1016/j.cell.2015.07.058
Viral and Cellular Genomes Activate Distinct DNA Damage Responses
Abstract
In response to cellular genome breaks, MRE11/RAD50/NBS1 (MRN) activates a global ATM DNA damage response (DDR) that prevents cellular replication. Here, we show that MRN-ATM also has critical functions in defending the cell against DNA viruses. We reveal temporally distinct responses to adenovirus genomes: a critical MRN-ATM DDR that must be inactivated by E1B-55K/E4-ORF3 viral oncoproteins and a global MRN-independent ATM DDR to viral nuclear domains that does not impact viral replication. We show that MRN binds to adenovirus genomes and activates a localized ATM response that specifically prevents viral DNA replication. In contrast to chromosomal breaks, ATM activation is not amplified by H2AX across megabases of chromatin to induce global signaling and replicative arrest. Thus, γH2AX foci discriminate "self" and "non-self" genomes and determine whether a localized anti-viral or global ATM response is appropriate. This provides an elegant mechanism to neutralize viral genomes without jeopardizing cellular viability.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
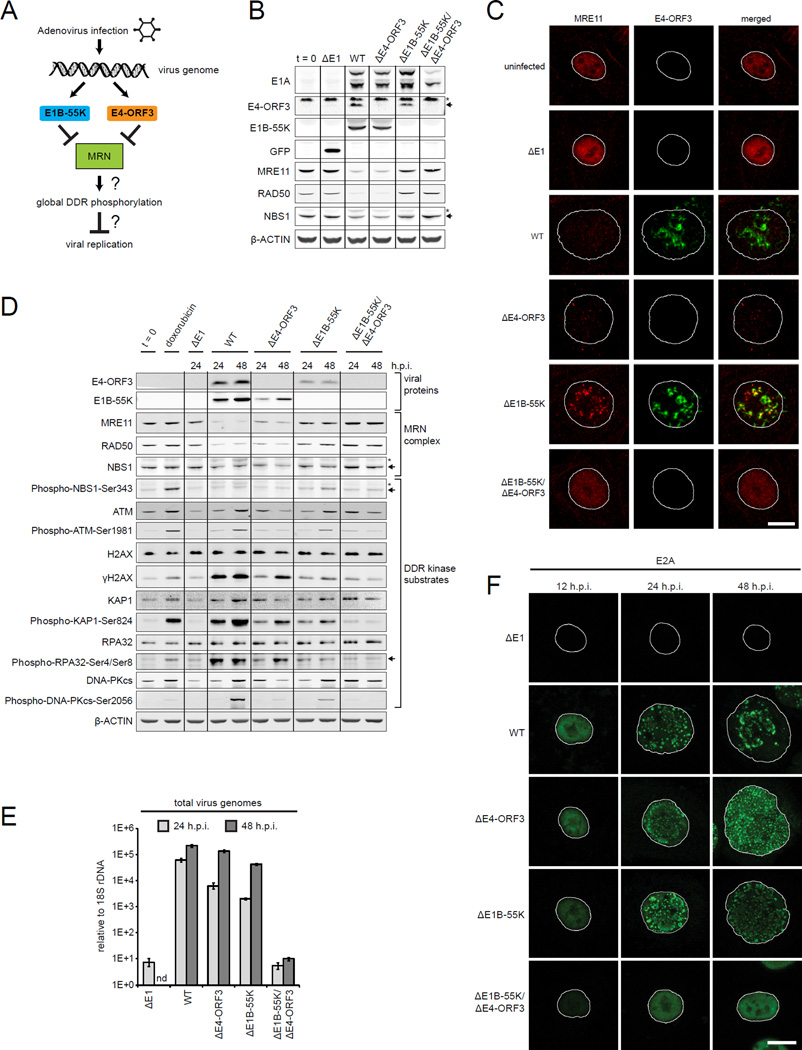
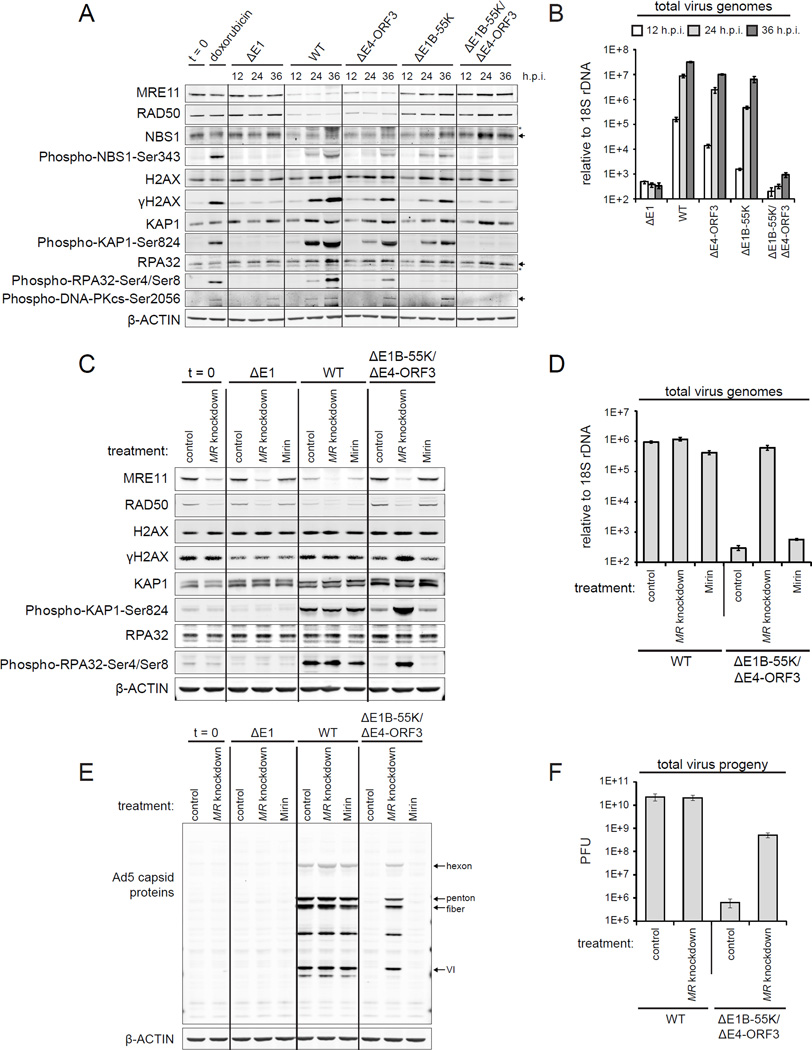
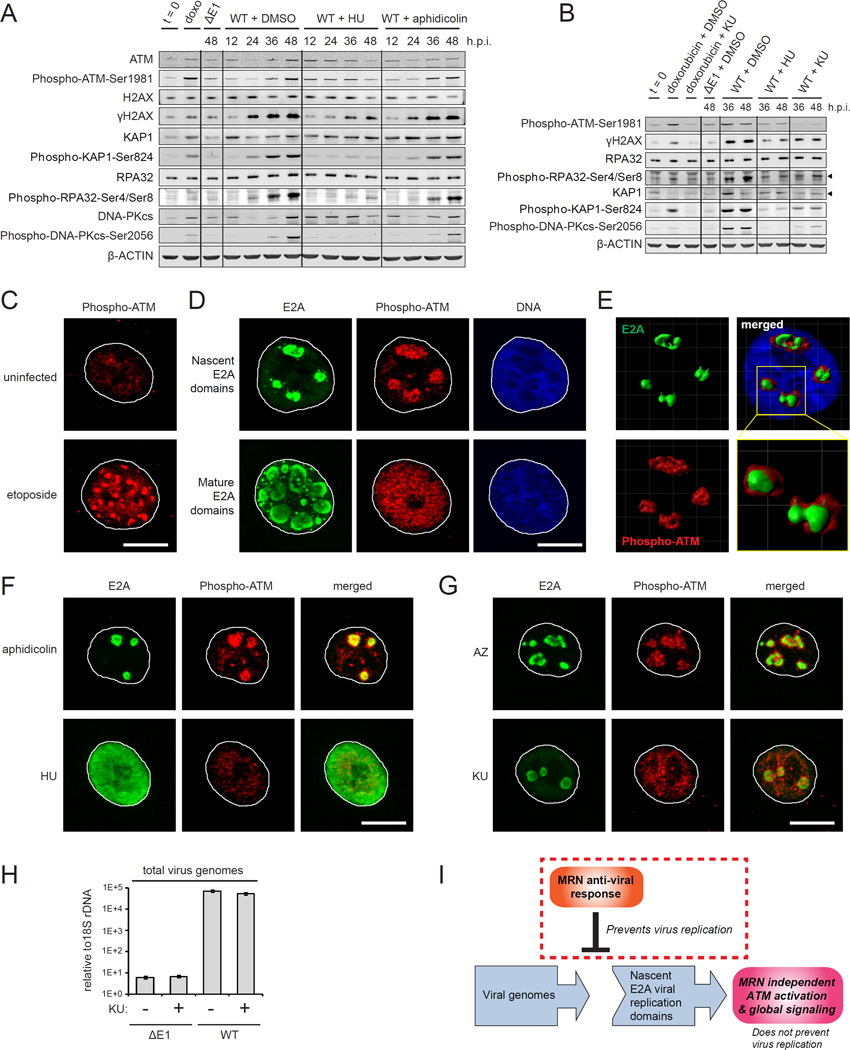
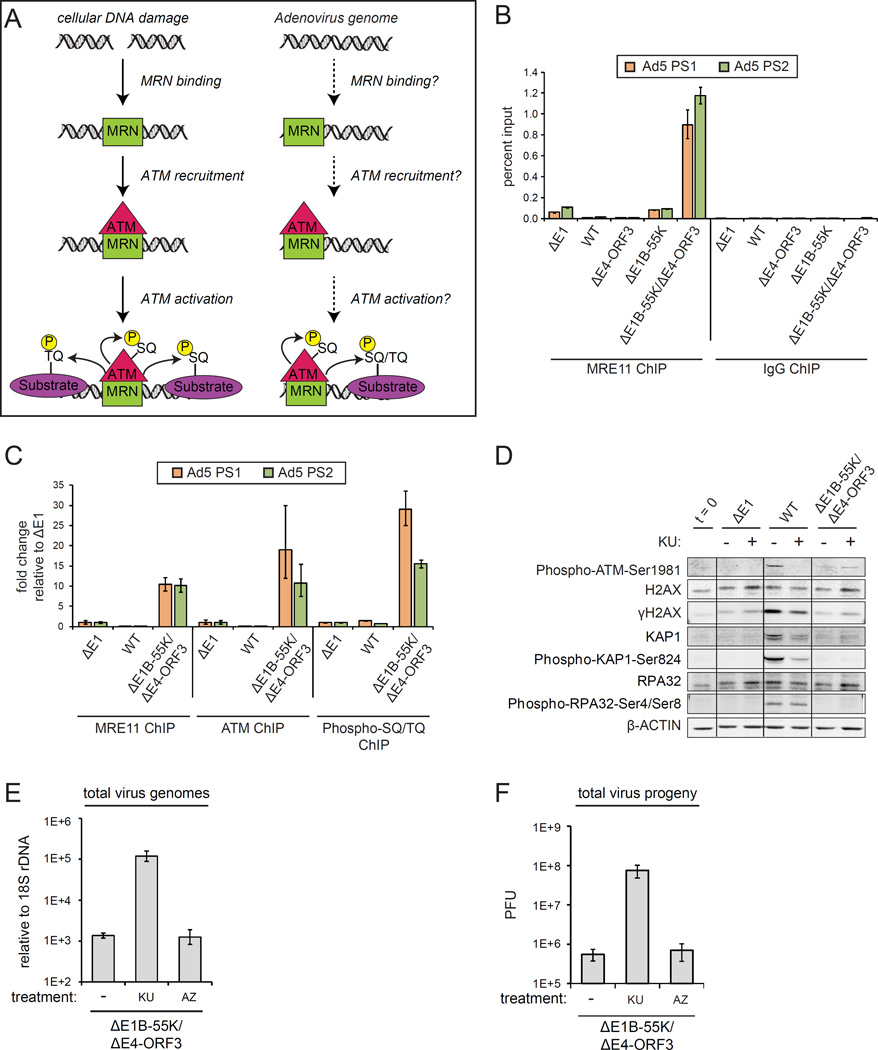
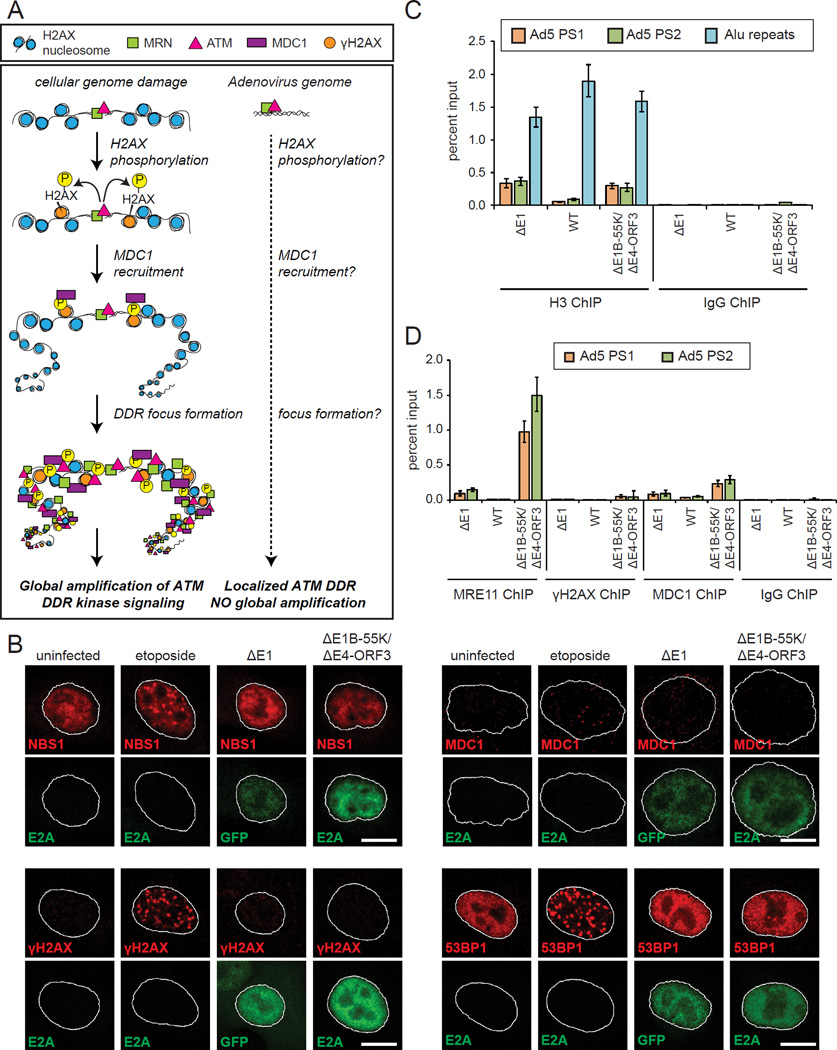
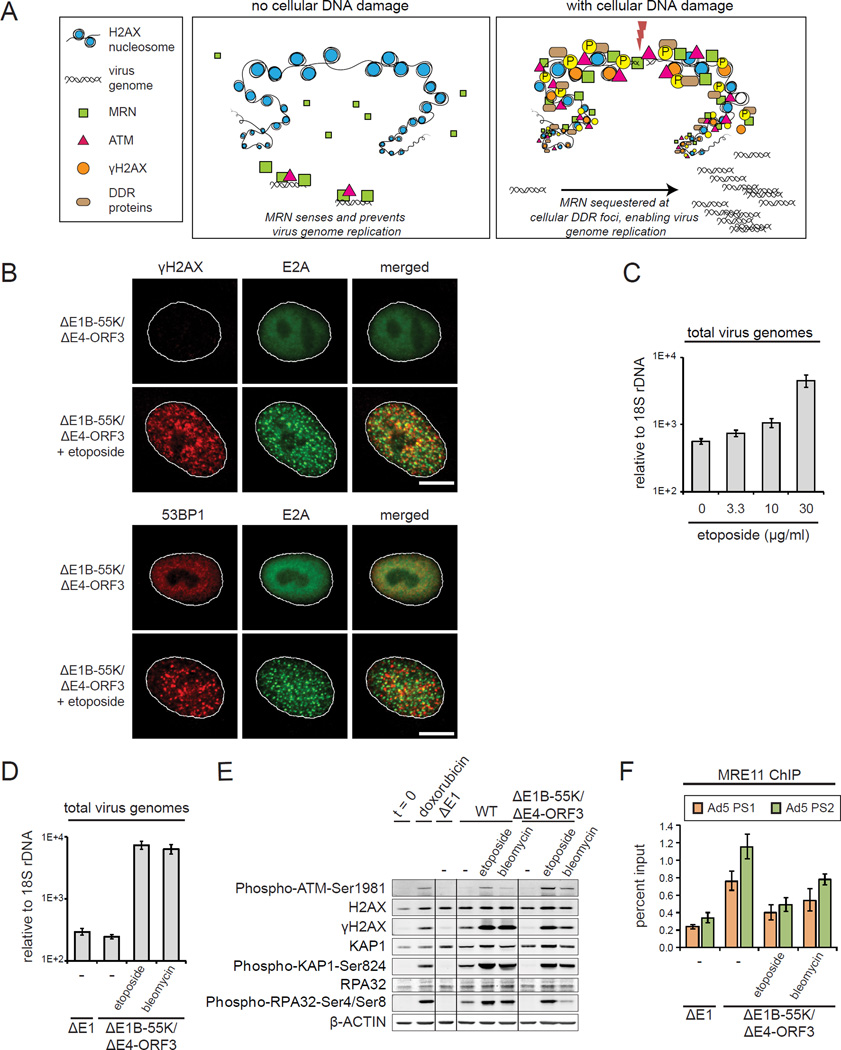
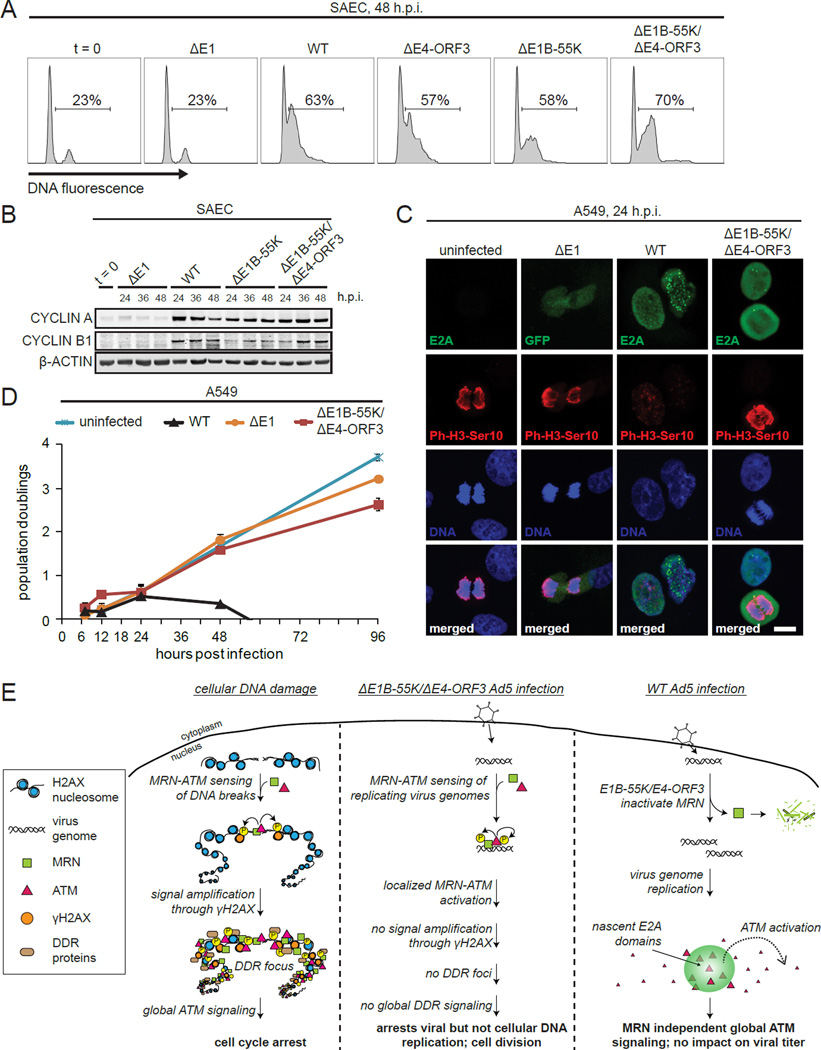
References
-
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. - PubMed
-
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8173–8178. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
