

Immune Mechanisms in Arterial Hypertension
- PMID: 26319245
- PMCID: PMC4769210
- DOI: 10.1681/ASN.2015050562
Immune Mechanisms in Arterial Hypertension
Abstract
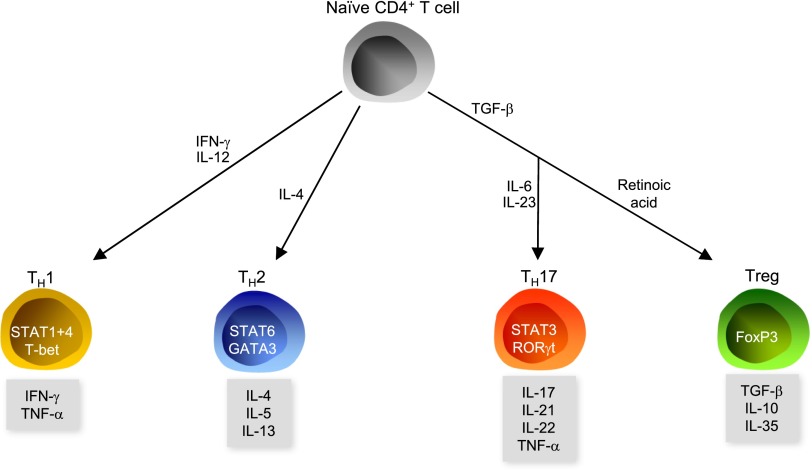
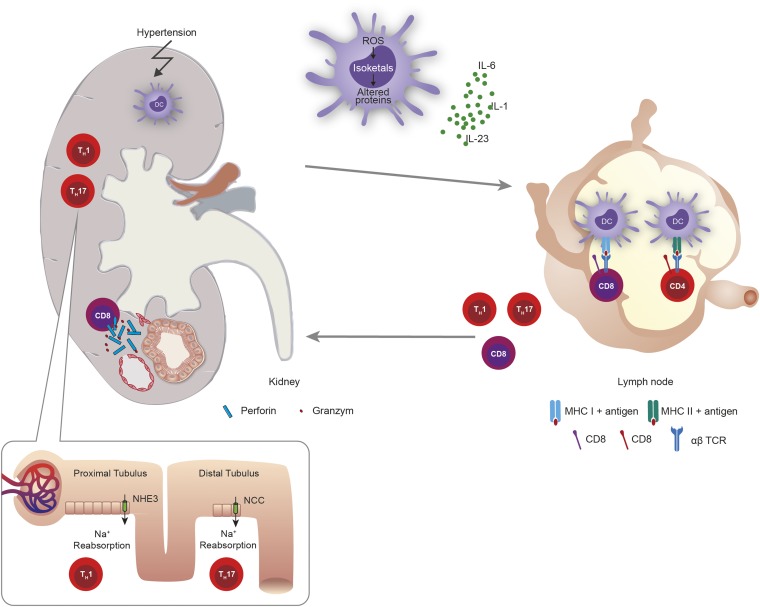
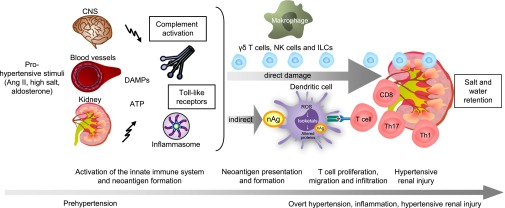
Traditionally, arterial hypertension and subsequent end-organ damage have been attributed to hemodynamic factors, but increasing evidence indicates that inflammation also contributes to the deleterious consequences of this disease. The immune system has evolved to prevent invasion of foreign organisms and to promote tissue healing after injury. However, this beneficial activity comes at a cost of collateral damage when the immune system overreacts to internal injury, such as prehypertension. Renal inflammation results in injury and impaired urinary sodium excretion, and vascular inflammation leads to endothelial dysfunction, increased vascular resistance, and arterial remodeling and stiffening. Notably, modulation of the immune response can reduce the severity of BP elevation and hypertensive end-organ damage in several animal models. Indeed, recent studies have improved our understanding of how the immune response affects the pathogenesis of arterial hypertension, but the remarkable advances in basic immunology made during the last few years still await translation to the field of hypertension. This review briefly summarizes recent advances in immunity and hypertension as well as hypertensive end-organ damage.
Keywords: adaptive immunity; dendritic cells; hypertension; innate immunity.
Copyright © 2016 by the American Society of Nephrology.
Figures
References
-
- Beilin LJ, Goldby FS, Mohring J: High arterial pressure versus humoral factors in the pathogenesis of the vascular lesions of malignant hypertension. Clin Sci Mol Med 52: 111–117, 1977 - PubMed
-
- Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ: Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
