

Matrix Metalloproteinases as Regulators of Vein Structure and Function: Implications in Chronic Venous Disease
- PMID: 26319699
- PMCID: PMC4658486
- DOI: 10.1124/jpet.115.227330
Matrix Metalloproteinases as Regulators of Vein Structure and Function: Implications in Chronic Venous Disease
Abstract
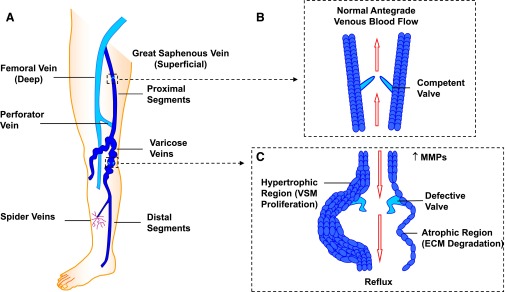
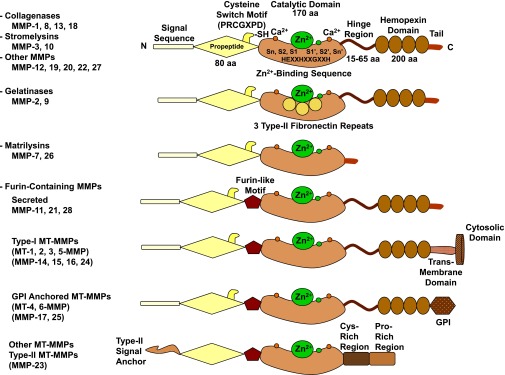
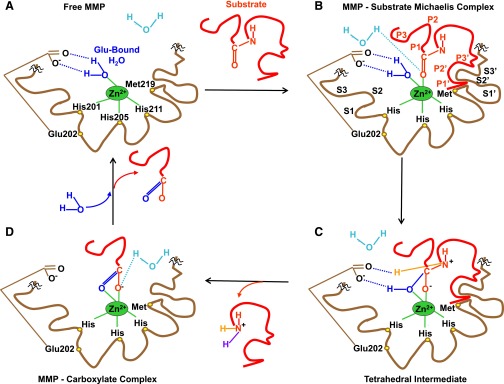
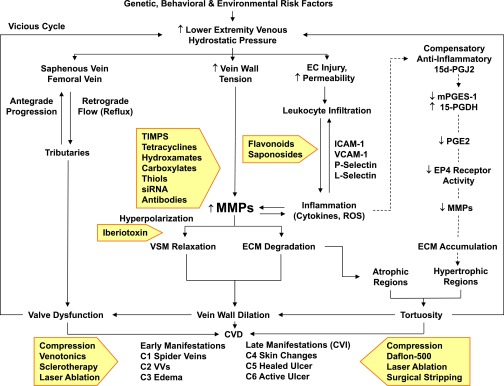
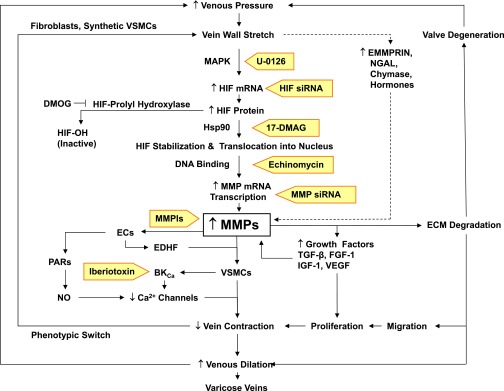
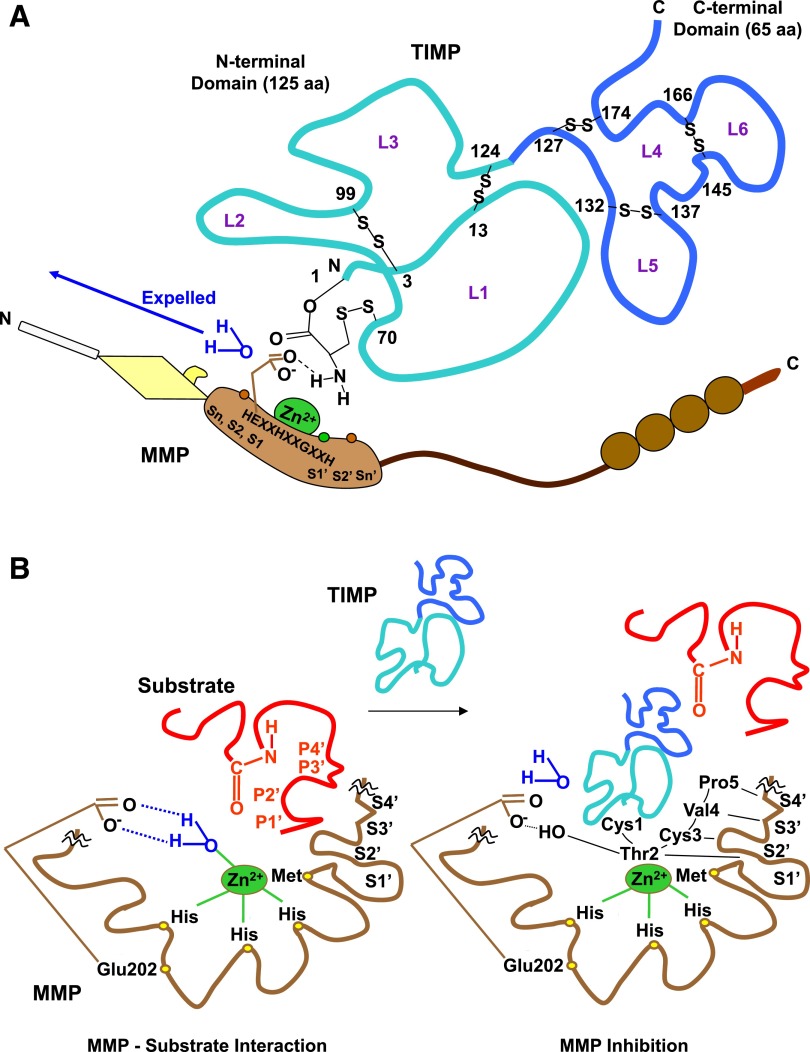
Lower-extremity veins have efficient wall structure and function and competent valves that permit upward movement of deoxygenated blood toward the heart against hydrostatic venous pressure. Matrix metalloproteinases (MMPs) play an important role in maintaining vein wall structure and function. MMPs are zinc-binding endopeptidases secreted as inactive pro-MMPs by fibroblasts, vascular smooth muscle (VSM), and leukocytes. Pro-MMPs are activated by various activators including other MMPs and proteinases. MMPs cause degradation of extracellular matrix (ECM) proteins such as collagen and elastin, and could have additional effects on the endothelium, as well as VSM cell migration, proliferation, Ca(2+) signaling, and contraction. Increased lower-extremity hydrostatic venous pressure is thought to induce hypoxia-inducible factors and other MMP inducers/activators such as extracellular matrix metalloproteinase inducer, prostanoids, chymase, and hormones, leading to increased MMP expression/activity, ECM degradation, VSM relaxation, and venous dilation. Leukocyte infiltration and inflammation of the vein wall cause further increases in MMPs, vein wall dilation, valve degradation, and different clinical stages of chronic venous disease (CVD), including varicose veins (VVs). VVs are characterized by ECM imbalance, incompetent valves, venous reflux, wall dilation, and tortuosity. VVs often show increased MMP levels, but may show no change or decreased levels, depending on the VV region (atrophic regions with little ECM versus hypertrophic regions with abundant ECM) and MMP form (inactive pro-MMP versus active MMP). Management of VVs includes compression stockings, venotonics, and surgical obliteration or removal. Because these approaches do not treat the causes of VVs, alternative methods are being developed. In addition to endogenous tissue inhibitors of MMPs, synthetic MMP inhibitors have been developed, and their effects in the treatment of VVs need to be examined.
Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
References
-
- Aimes RT, Quigley JP. (1995) Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J Biol Chem 270:5872–5876. - PubMed
-
- Aravind B, Saunders B, Navin T, Sandison A, Monaco C, Paleolog EM, Davies AH. (2010) Inhibitory effect of TIMP influences the morphology of varicose veins. Eur J Vasc Endovasc Surg 40:754–765. - PubMed
-
- Aremu MA, Mahendran B, Butcher W, Khan Z, Colgan MP, Moore DJ, Madhavan P, Shanik DG. (2004) Prospective randomized controlled trial: conventional versus powered phlebectomy. J Vasc Surg 39:88–94. - PubMed
-
- Asanuma K, Magid R, Johnson C, Nerem RM, Galis ZS. (2003) Uniaxial strain upregulates matrix-degrading enzymes produced by human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 284:H1778–H1784. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
