

Glycogen synthase kinase-3β modulation of glucocorticoid responsiveness in COPD
- PMID: 26320152
- PMCID: PMC4652154
- DOI: 10.1152/ajplung.00077.2015
Glycogen synthase kinase-3β modulation of glucocorticoid responsiveness in COPD
Abstract
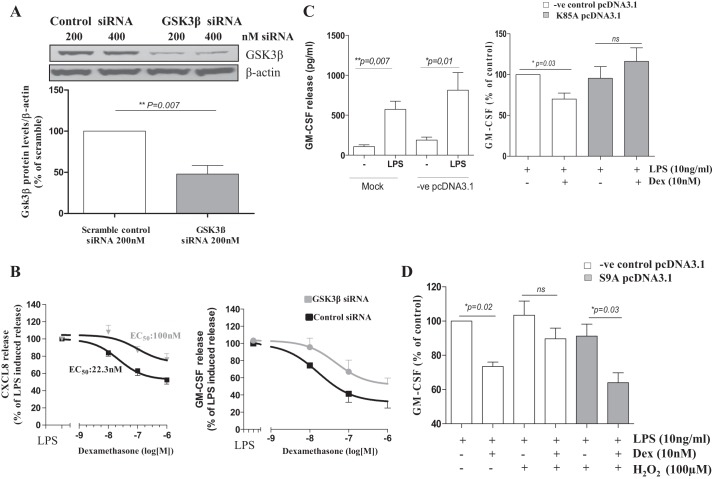
In chronic obstructive pulmonary disease (COPD), oxidative stress regulates the inflammatory response of bronchial epithelium and monocytes/macrophages through kinase modulation and has been linked to glucocorticoid unresponsiveness. Glycogen synthase-3β (GSK3β) inactivation plays a key role in mediating signaling processes upon reactive oxygen species (ROS) exposure. We hypothesized that GSK3β is involved in oxidative stress-induced glucocorticoid insensitivity in COPD. We studied levels of phospho-GSK3β-Ser9, a marker of GSK3β inactivation, in lung sections and cultured monocytes and bronchial epithelial cells of COPD patients, control smokers, and nonsmokers. We observed increased levels of phospho-GSK3β-Ser9 in monocytes, alveolar macrophages, and bronchial epithelial cells from COPD patients and control smokers compared with nonsmokers. Pharmacological inactivation of GSK3β did not affect CXCL8 or granulocyte-macrophage colony-stimulating factor (GM-CSF) expression but resulted in glucocorticoid insensitivity in vitro in both inflammatory and structural cells. Further mechanistic studies in monocyte and bronchial epithelial cell lines showed that GSK3β inactivation is a common effector of oxidative stress-induced activation of the MEK/ERK-1/2 and phosphatidylinositol 3-kinase/Akt signaling pathways leading to glucocorticoid unresponsiveness. In primary monocytes, the mechanism involved modulation of histone deacetylase 2 (HDAC2) activity in response to GSK3β inactivation. In conclusion, we demonstrate for the first time that ROS-induced glucocorticoid unresponsiveness in COPD is mediated through GSK3β, acting as a ROS-sensitive hub.
Keywords: COPD; epithelial cells; inflammatory responses; monocytes; oxidative stress.
Copyright © 2015 the American Physiological Society.
Figures






References
-
- Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet 373: 1905–1917, 2009. - PubMed
-
- Barnes PJ, Ito K, Adcock IM. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet 363: 731–733, 2004. - PubMed
-
- Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 131: 636–645, 2013. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous