

Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved β Cell Function
- PMID: 26321660
- PMCID: PMC4598285
- DOI: 10.1016/j.cmet.2015.07.025
Aging-Dependent Demethylation of Regulatory Elements Correlates with Chromatin State and Improved β Cell Function
Abstract
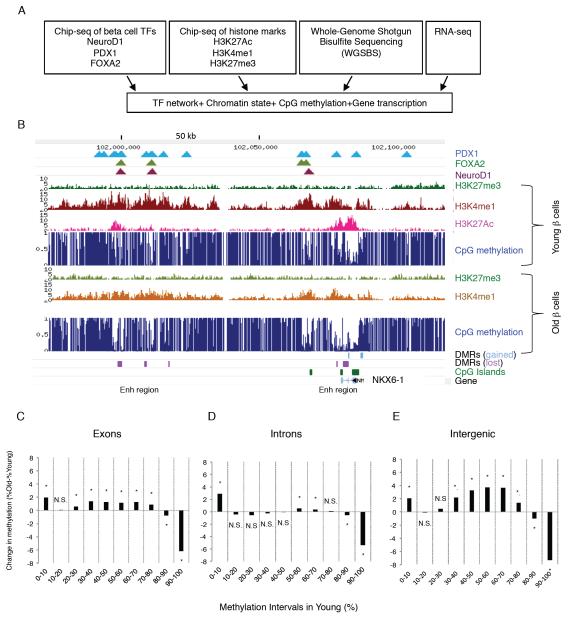
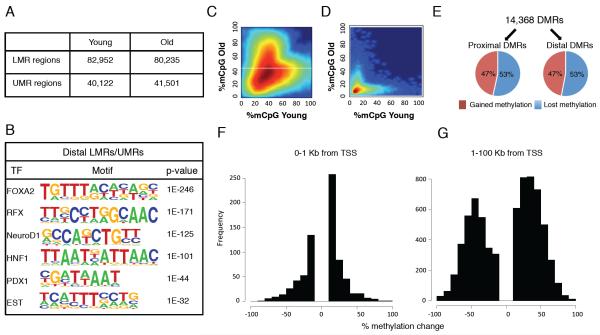
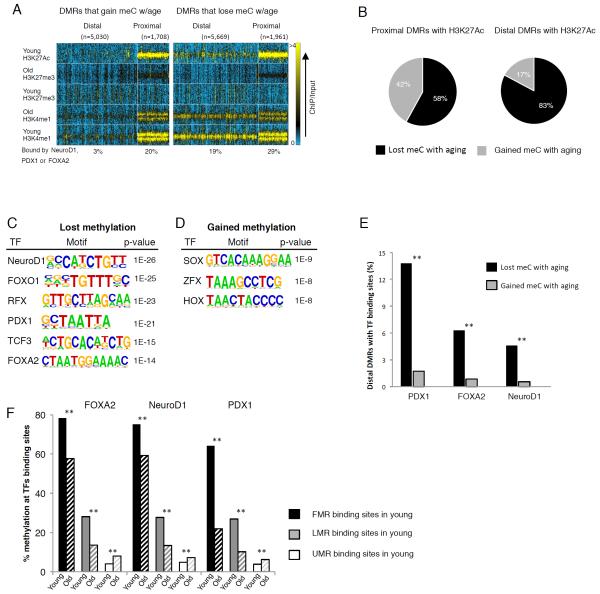
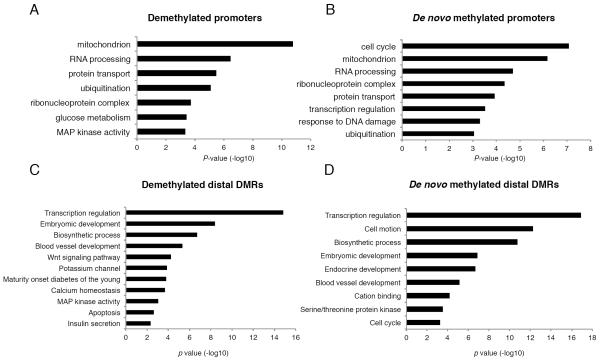
Aging is driven by changes of the epigenetic state that are only partially understood. We performed a comprehensive epigenomic analysis of the pancreatic β cell, key player in glucose homeostasis, in adolescent and very old mice. We observe a global methylation drift resulting in an overall more leveled methylome in old β cells. Importantly, we discover targeted changes in the methylation status of β cell proliferation and function genes that go against the global methylation drift, are specific to β cells, and correlate with repression of the proliferation program and activation of metabolic regulators. These targeted alterations are associated with specific chromatin marks and transcription factor occupancy in young β cells. Strikingly, we find β cell function improved in aged mice, as predicted by the changes in methylome and transcriptome. Thus, aging of terminally differentiated cells in mammals is not always coupled to functional decline.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
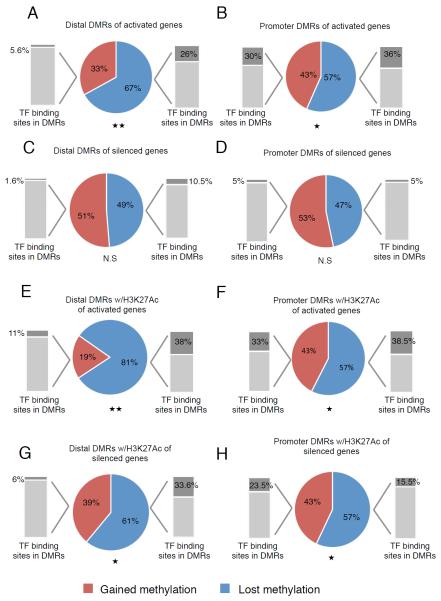
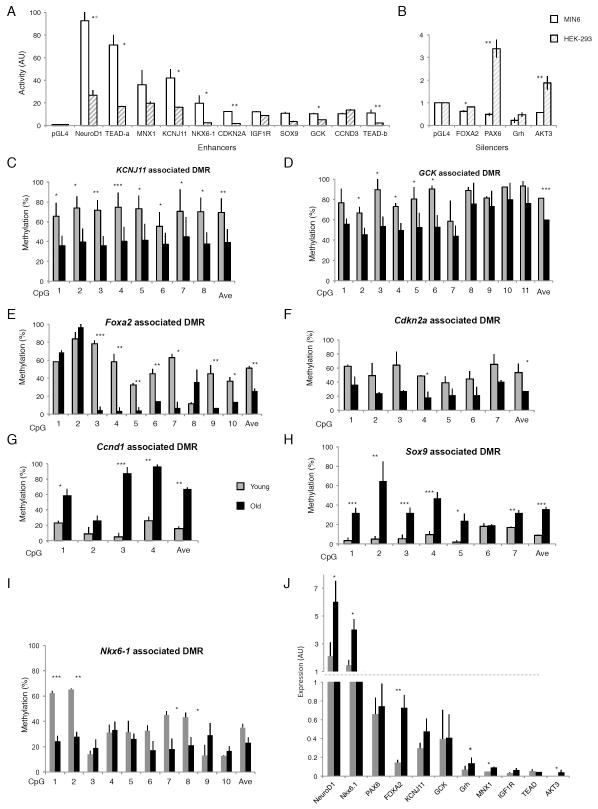
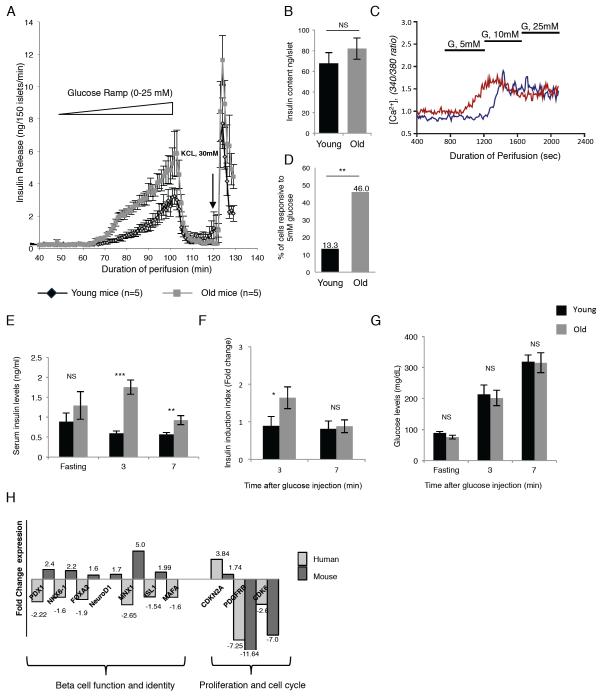
References
-
- Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. Journal of molecular endocrinology. 2007;38:193–206. - PubMed
-
- Berninger P, Gaidatzis D, van Nimwegen E, Zavolan M. Computational analysis of small RNA cloning data. Methods. 2008;44:13–21. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6–21. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
