

Oncolytic viruses: a new class of immunotherapy drugs
- PMID: 26323545
- PMCID: PMC7097180
- DOI: 10.1038/nrd4663
Oncolytic viruses: a new class of immunotherapy drugs
Erratum in
-
Oncolytic viruses: a new class of immunotherapy drugs.Nat Rev Drug Discov. 2016 Aug 30;15(9):660. doi: 10.1038/nrd.2016.178. Nat Rev Drug Discov. 2016. PMID: 30907381 Free PMC article.
Abstract
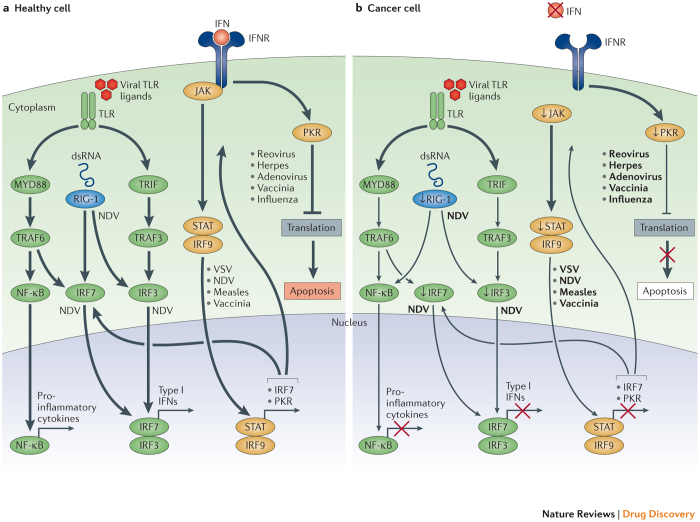
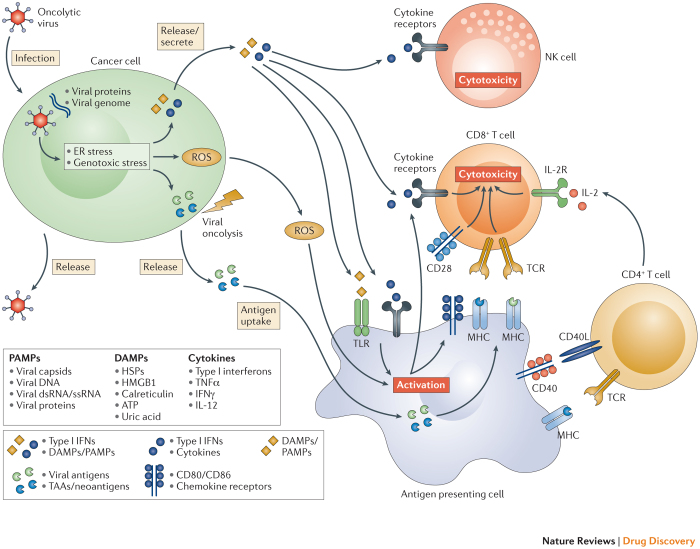
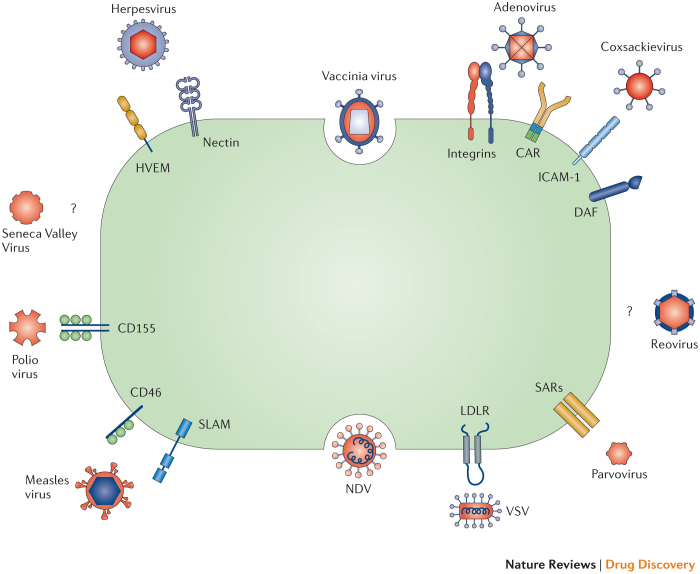
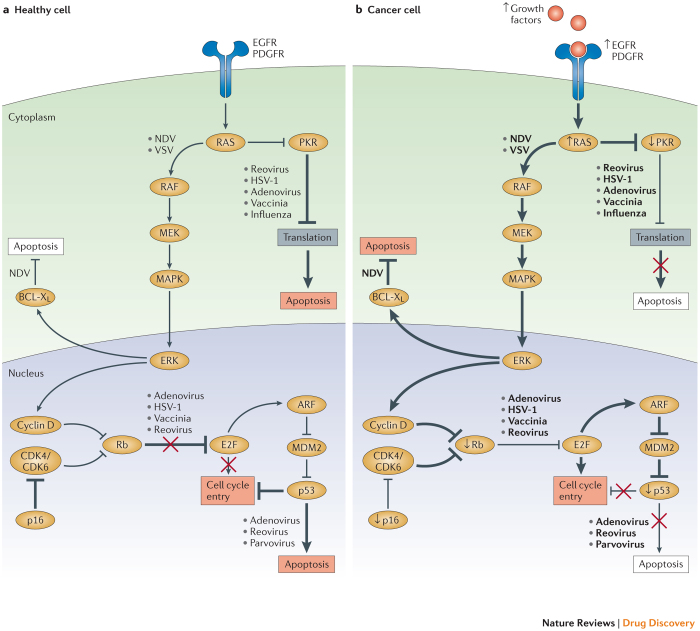
Oncolytic viruses represent a new class of therapeutic agents that promote anti-tumour responses through a dual mechanism of action that is dependent on selective tumour cell killing and the induction of systemic anti-tumour immunity. The molecular and cellular mechanisms of action are not fully elucidated but are likely to depend on viral replication within transformed cells, induction of primary cell death, interaction with tumour cell antiviral elements and initiation of innate and adaptive anti-tumour immunity. A variety of native and genetically modified viruses have been developed as oncolytic agents, and the approval of the first oncolytic virus by the US Food and Drug Administration (FDA) is anticipated in the near future. This Review provides a comprehensive overview of the basic biology supporting oncolytic viruses as cancer therapeutic agents, describes oncolytic viruses in advanced clinical trials and discusses the unique challenges in the development of oncolytic viruses as a new class of drugs for the treatment of cancer.
Conflict of interest statement
H.L.K. serves as a consultant for Amgen and has received honoraria. The other authors have no competing interests to disclose.
Figures
References
-
- Moore AE. The destructive effect of the virus of Russian Far East encephalitis on the transplantable mouse sarcoma 180. Cancer. 1949;2:525–534. - PubMed
-
- Moore AE. Effect of inoculation of the viruses of influenza A and herpes simplex on the growth of transplantable tumors in mice. Cancer. 1949;2:516–524. - PubMed
-
- Andtbacka, R. H. et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol.10.1200/JCO.2014.58.3377 (2015). Phase III clinical trial that led to the pending FDA approval of T-VEC for clinical use in the United States for melanoma. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
