

Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death
- PMID: 26323688
- PMCID: PMC4555813
- DOI: 10.1083/jcb.201503044
Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death
Erratum in
-
Correction: Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death.J Cell Biol. 2016 Mar 28;212(7):881-2. doi: 10.1083/jcb.20150304403082016c. Epub 2016 Mar 21. J Cell Biol. 2016. PMID: 27002169 Free PMC article. No abstract available.
Abstract
Hexokinase II (HK2), a key enzyme involved in glucose metabolism, is regulated by growth factor signaling and is required for initiation and maintenance of tumors. Here we show that metabolic stress triggered by perturbation of receptor tyrosine kinase FLT3 in non-acute myeloid leukemia cells sensitizes cancer cells to autophagy inhibition and leads to excessive activation of chaperone-mediated autophagy (CMA). Our data demonstrate that FLT3 is an important sensor of cellular nutritional state and elucidate the role and molecular mechanism of CMA in metabolic regulation and mediating cancer cell death. Importantly, our proteome analysis revealed that HK2 is a CMA substrate and that its degradation by CMA is regulated by glucose availability. We reveal a new mechanism by which excessive activation of CMA may be exploited pharmacologically to eliminate cancer cells by inhibiting both FLT3 and autophagy. Our study delineates a novel pharmacological strategy to promote the degradation of HK2 in cancer cells.
© 2015 Xia et al.
Figures
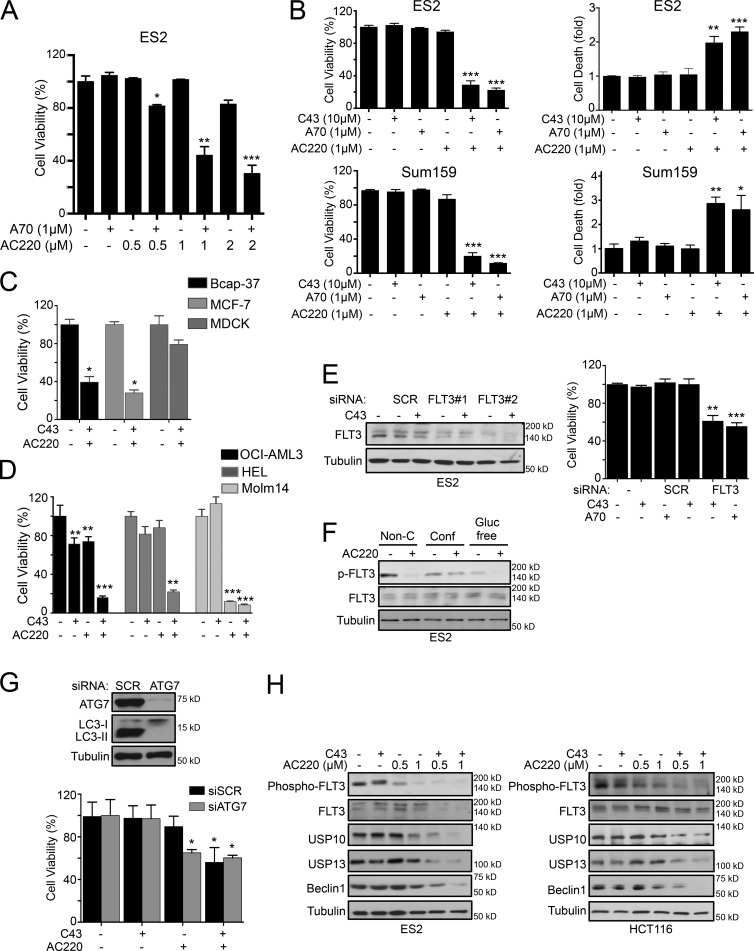
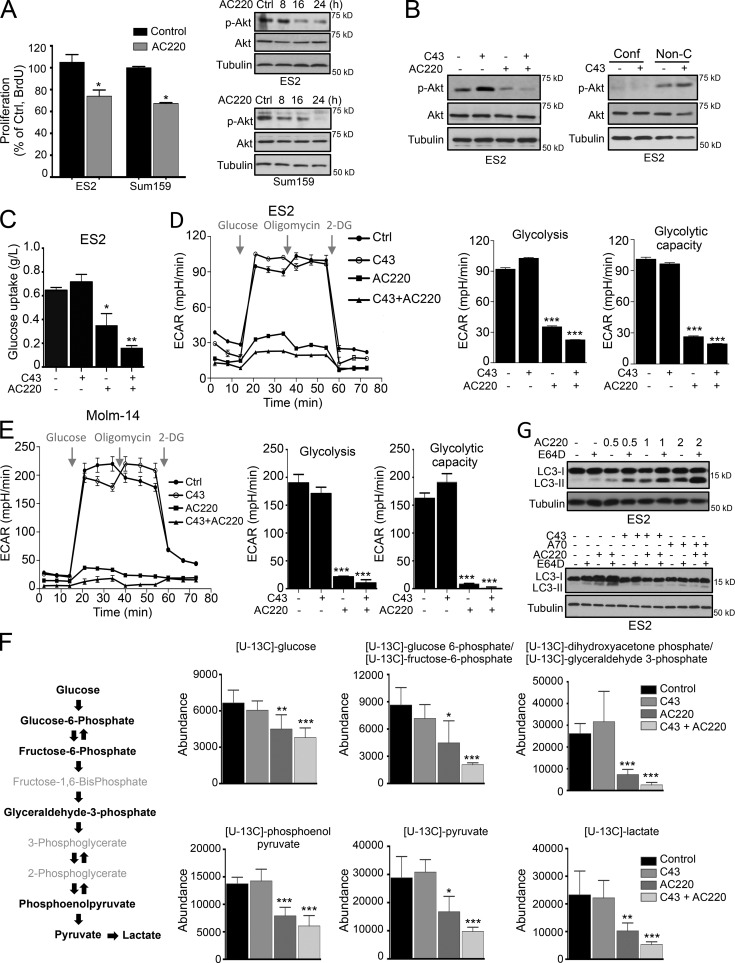
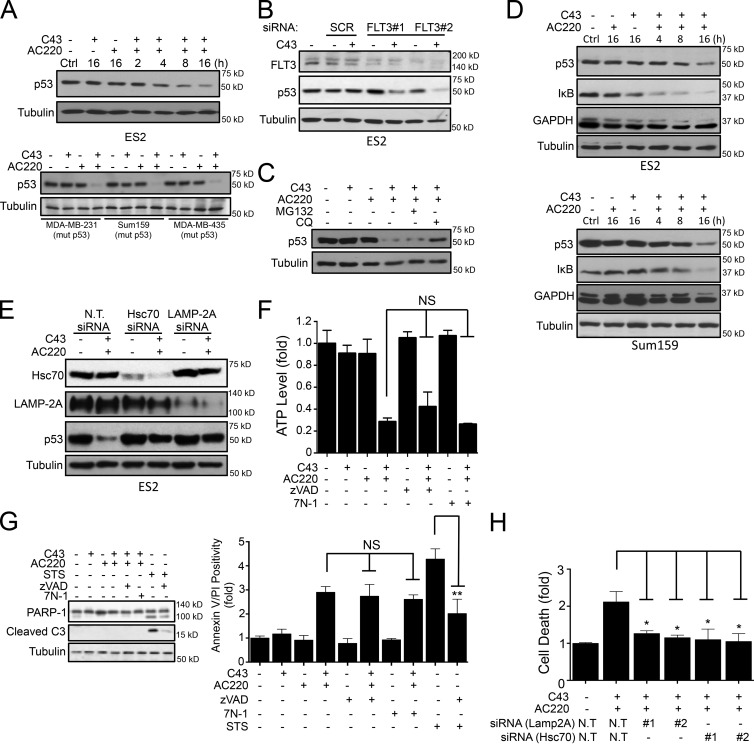
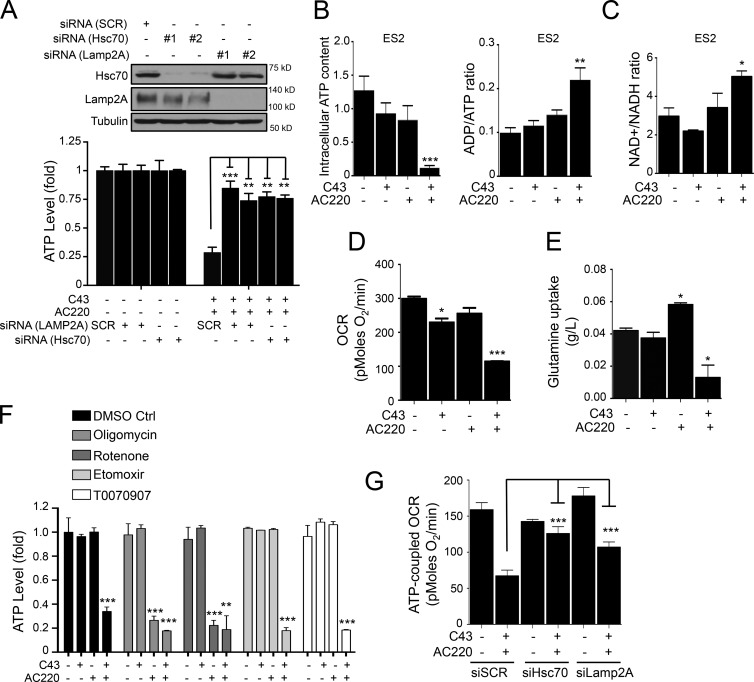
Comment in
-
Activation of chaperone-mediated autophagy as a potential anticancer therapy.Autophagy. 2015;11(12):2370-1. doi: 10.1080/15548627.2015.1106666. Autophagy. 2015. PMID: 26577179 Free PMC article.
References
-
- Aniento F., Roche E., Cuervo A.M., and Knecht E.. 1993. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 268:10463–10470. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
