

PPM1G Binds 7SK RNA and Hexim1 To Block P-TEFb Assembly into the 7SK snRNP and Sustain Transcription Elongation
- PMID: 26324325
- PMCID: PMC4609742
- DOI: 10.1128/MCB.00226-15
PPM1G Binds 7SK RNA and Hexim1 To Block P-TEFb Assembly into the 7SK snRNP and Sustain Transcription Elongation
Abstract
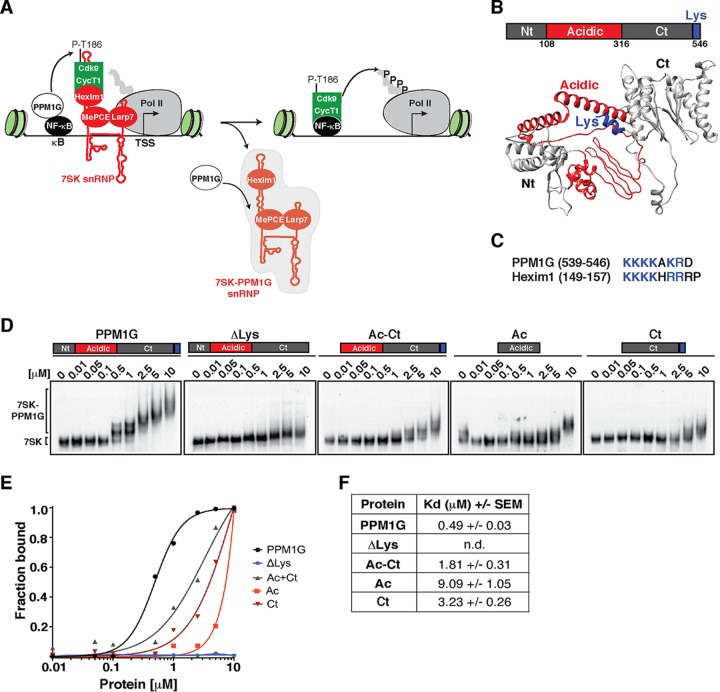
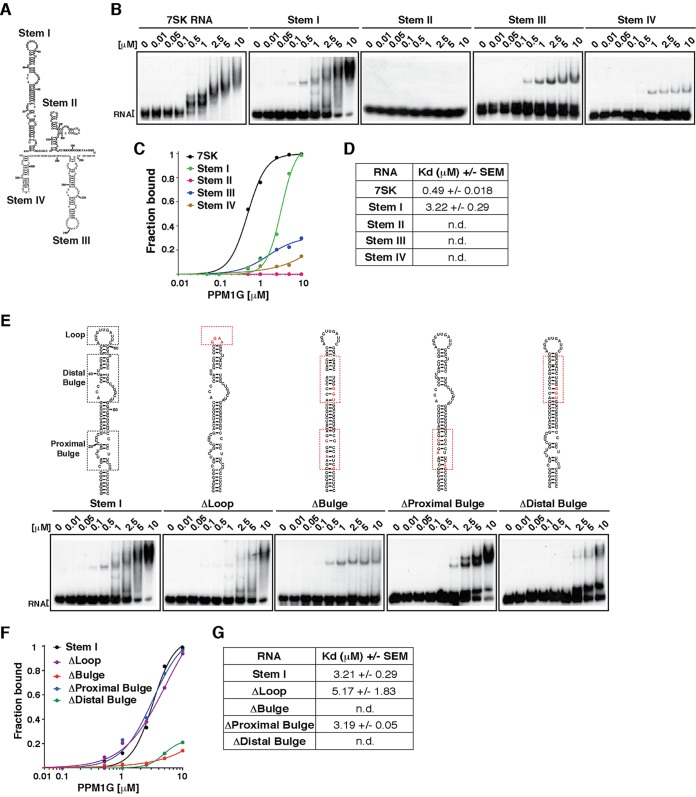
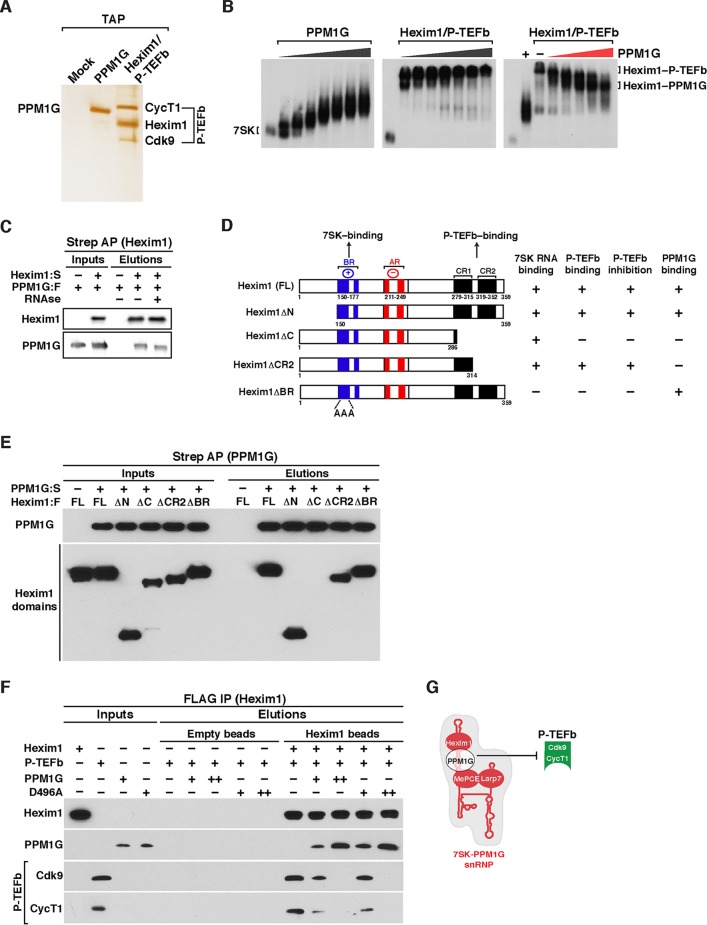
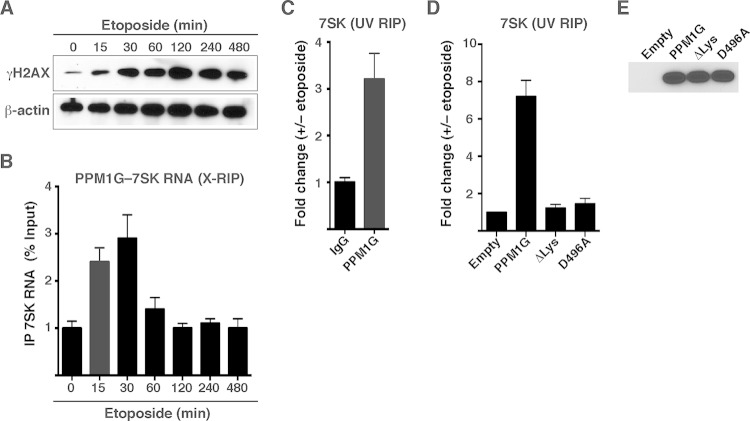
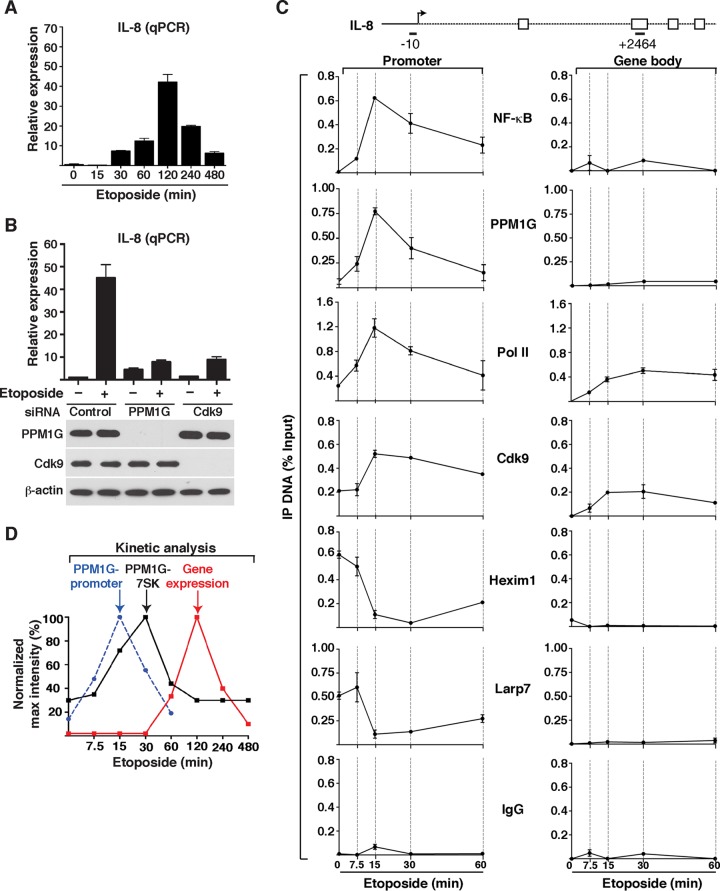
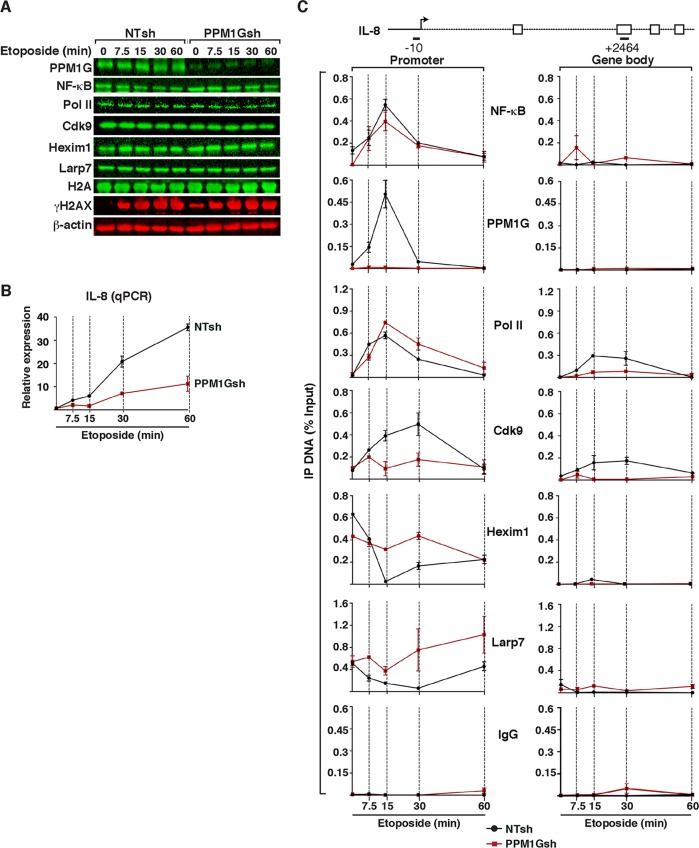
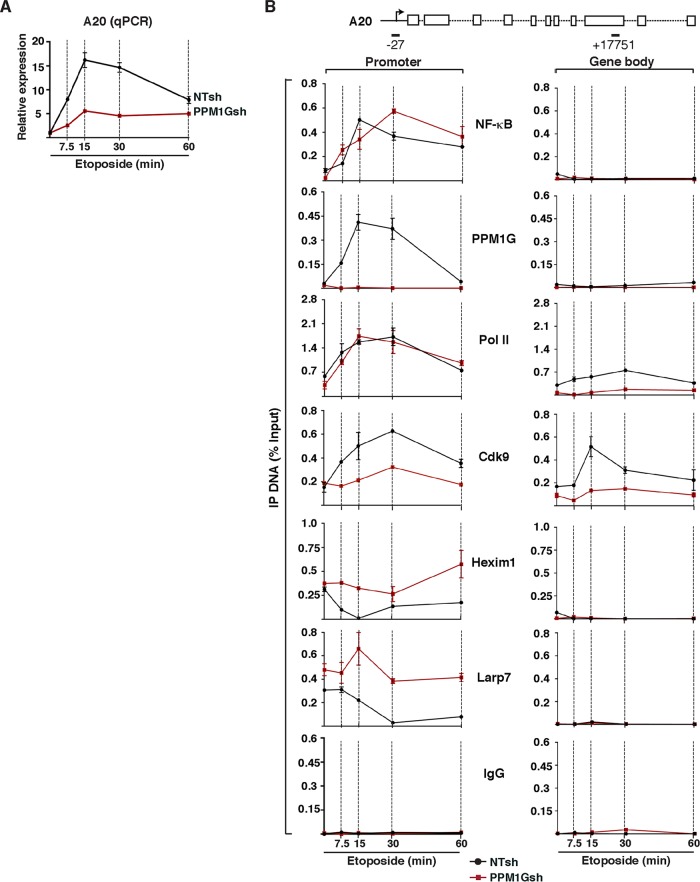
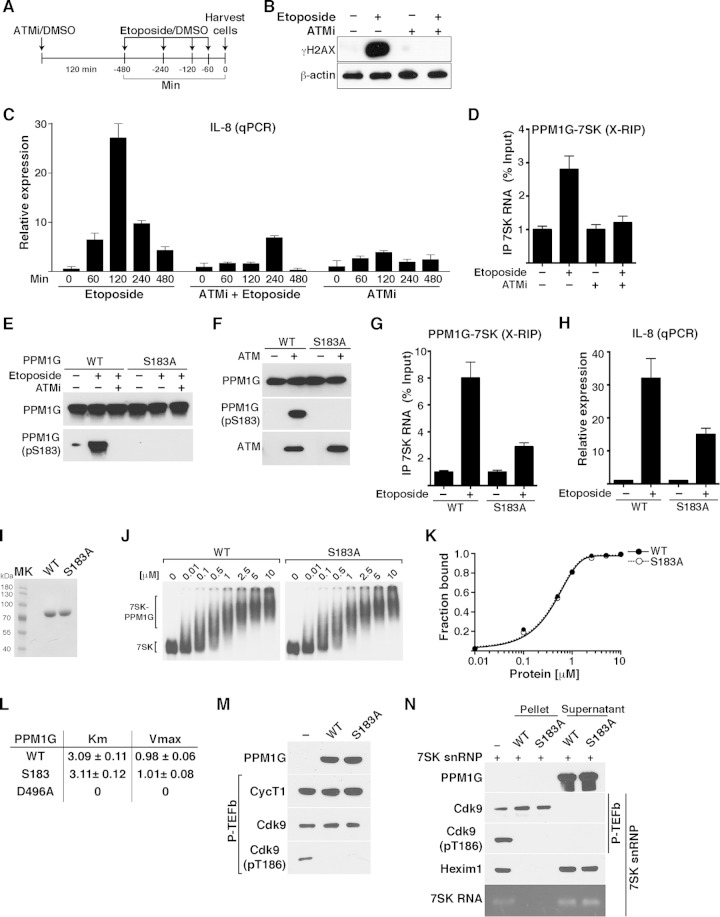
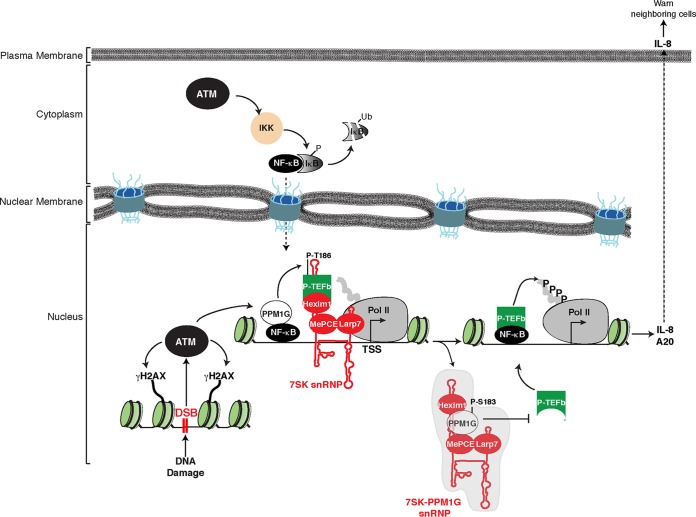
Transcription elongation programs are vital for the precise regulation of several biological processes. One key regulator of such programs is the P-TEFb kinase, which phosphorylates RNA polymerase II (Pol II) once released from the inhibitory 7SK small nuclear ribonucleoprotein (snRNP) complex. Although mechanisms of P-TEFb release from the snRNP are becoming clearer, how P-TEFb remains in the 7SK-unbound state to sustain transcription elongation programs remains unknown. Here we report that the PPM1G phosphatase (inducibly recruited by nuclear factor κB [NF-κB] to target promoters) directly binds 7SK RNA and the kinase inhibitor Hexim1 once P-TEFb has been released from the 7SK snRNP. This dual binding activity of PPM1G blocks P-TEFb reassembly onto the snRNP to sustain NF-κB-mediated Pol II transcription in response to DNA damage. Notably, the PPM1G-7SK RNA interaction is direct, kinetically follows the recruitment of PPM1G to promoters to activate NF-κB transcription, and is reversible, since the complex disassembles before resolution of the program. Strikingly, we found that the ataxia telangiectasia mutated (ATM) kinase regulates the interaction between PPM1G and the 7SK snRNP through site-specific PPM1G phosphorylation. The precise and temporally regulated interaction of a cellular enzyme and a noncoding RNA provides a new paradigm for simultaneously controlling the activation and maintenance of inducible transcription elongation programs.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation.Cell Rep. 2013 Dec 12;5(5):1256-68. doi: 10.1016/j.celrep.2013.11.003. Epub 2013 Dec 5. Cell Rep. 2013. PMID: 24316072 Free PMC article.
-
The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK.PLoS One. 2010 Aug 23;5(8):e12335. doi: 10.1371/journal.pone.0012335. PLoS One. 2010. PMID: 20808803 Free PMC article.
-
Release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein (snRNP) activates hexamethylene bisacetamide-inducible protein (HEXIM1) transcription.J Biol Chem. 2014 Apr 4;289(14):9918-25. doi: 10.1074/jbc.M113.539015. Epub 2014 Feb 10. J Biol Chem. 2014. PMID: 24515107 Free PMC article.
-
Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb.Nucleic Acids Res. 2016 Sep 19;44(16):7527-39. doi: 10.1093/nar/gkw585. Epub 2016 Jul 1. Nucleic Acids Res. 2016. PMID: 27369380 Free PMC article. Review.
-
Transcription elongation control by the 7SK snRNP complex: Releasing the pause.Cell Cycle. 2016 Aug 17;15(16):2115-2123. doi: 10.1080/15384101.2016.1181241. Epub 2016 May 6. Cell Cycle. 2016. PMID: 27152730 Free PMC article. Review.
Cited by
-
Detailed secondary structure models of invertebrate 7SK RNAs.RNA Biol. 2018 Feb 1;15(2):158-164. doi: 10.1080/15476286.2017.1412913. Epub 2017 Dec 21. RNA Biol. 2018. PMID: 29219696 Free PMC article.
-
Non-epigenetic induction of HEXIM1 by DNMT1 inhibitors and functional relevance.Sci Rep. 2020 Dec 3;10(1):21015. doi: 10.1038/s41598-020-78058-y. Sci Rep. 2020. PMID: 33273553 Free PMC article.
-
The phospho-landscape of the survival of motoneuron protein (SMN) protein: relevance for spinal muscular atrophy (SMA).Cell Mol Life Sci. 2022 Aug 25;79(9):497. doi: 10.1007/s00018-022-04522-9. Cell Mol Life Sci. 2022. PMID: 36006469 Free PMC article. Review.
-
Prognostic risk assessment model for alternative splicing events and splicing factors in malignant pleural mesothelioma.Cancer Med. 2023 Feb;12(4):4895-4906. doi: 10.1002/cam4.5174. Epub 2022 Aug 28. Cancer Med. 2023. PMID: 36031798 Free PMC article.
-
7SKiing on chromatin: Move globally, act locally.RNA Biol. 2016 Jun 2;13(6):545-53. doi: 10.1080/15476286.2016.1181254. Epub 2016 Apr 29. RNA Biol. 2016. PMID: 27128603 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous