

NOS1-derived nitric oxide promotes NF-κB transcriptional activity through inhibition of suppressor of cytokine signaling-1
- PMID: 26324446
- PMCID: PMC4577833
- DOI: 10.1084/jem.20140654
NOS1-derived nitric oxide promotes NF-κB transcriptional activity through inhibition of suppressor of cytokine signaling-1
Abstract
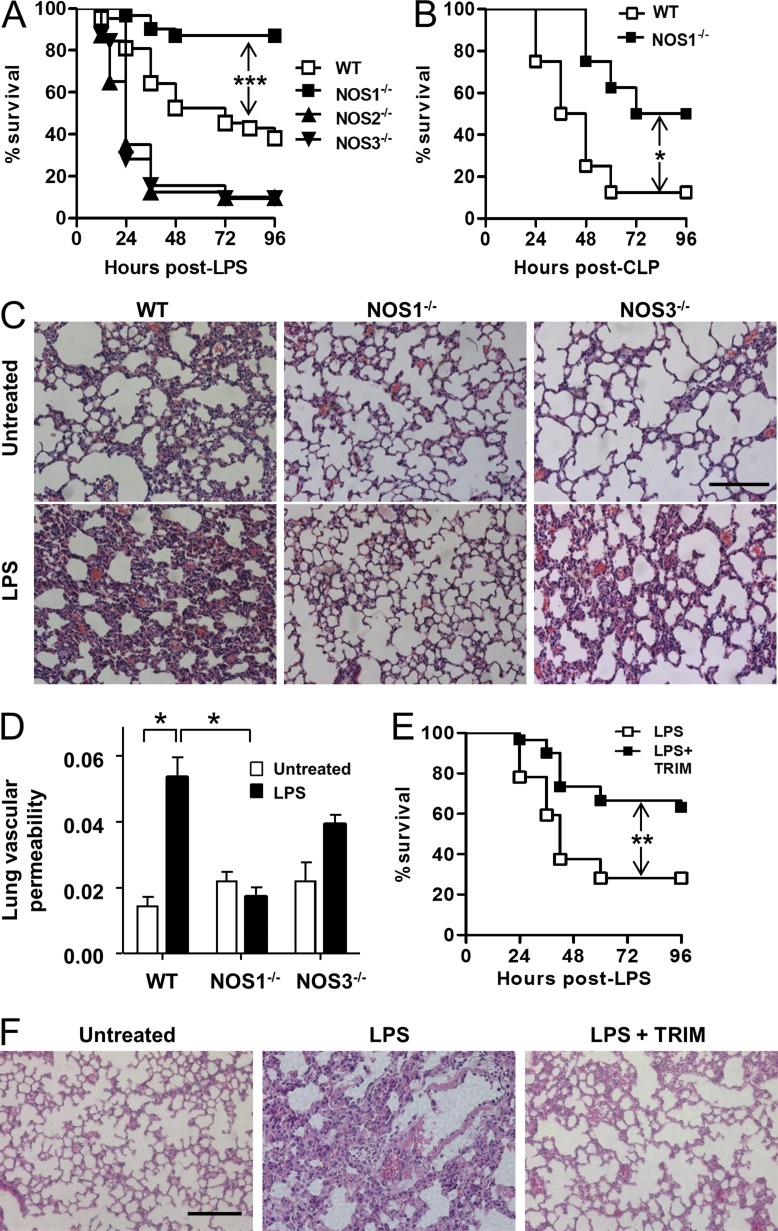
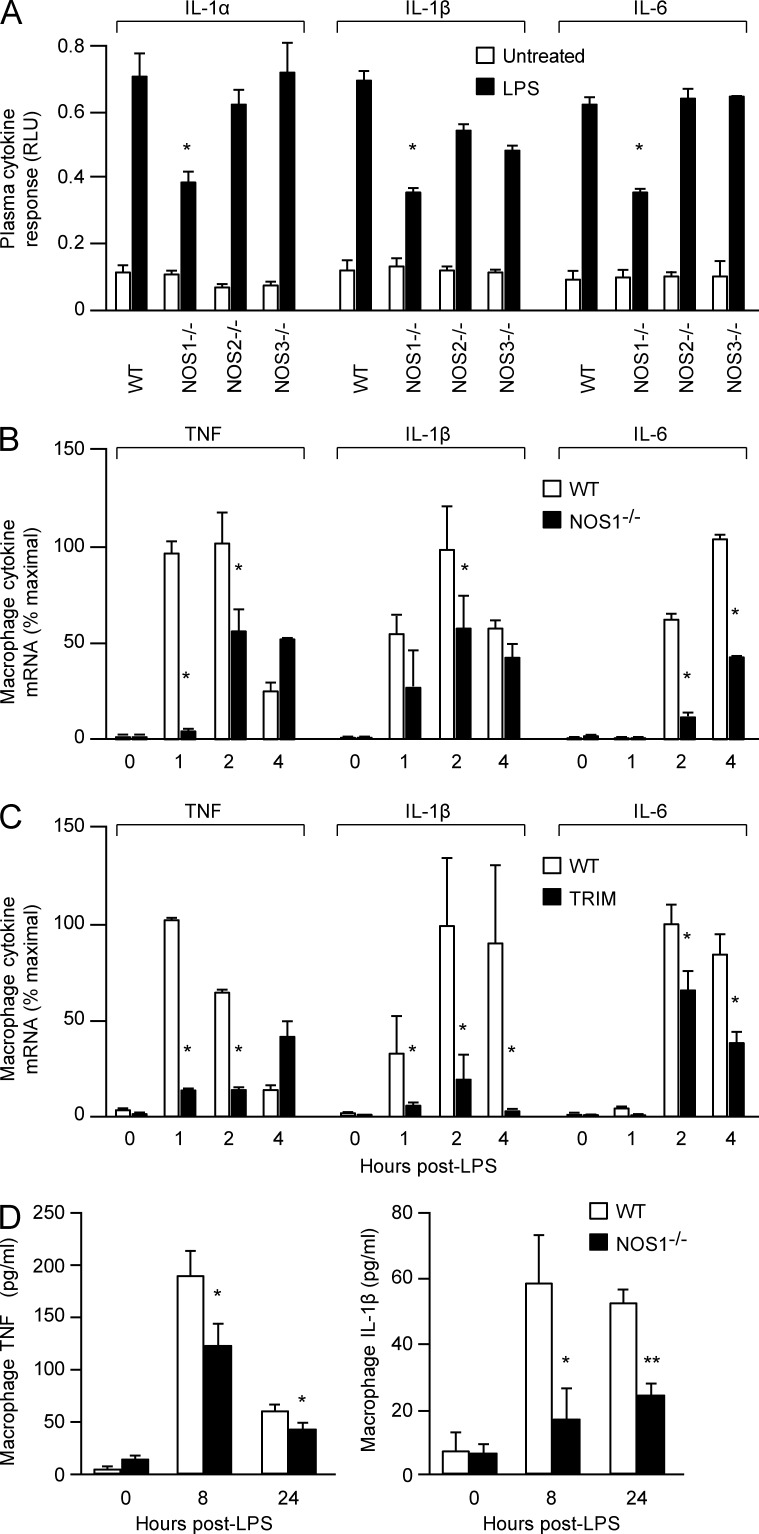
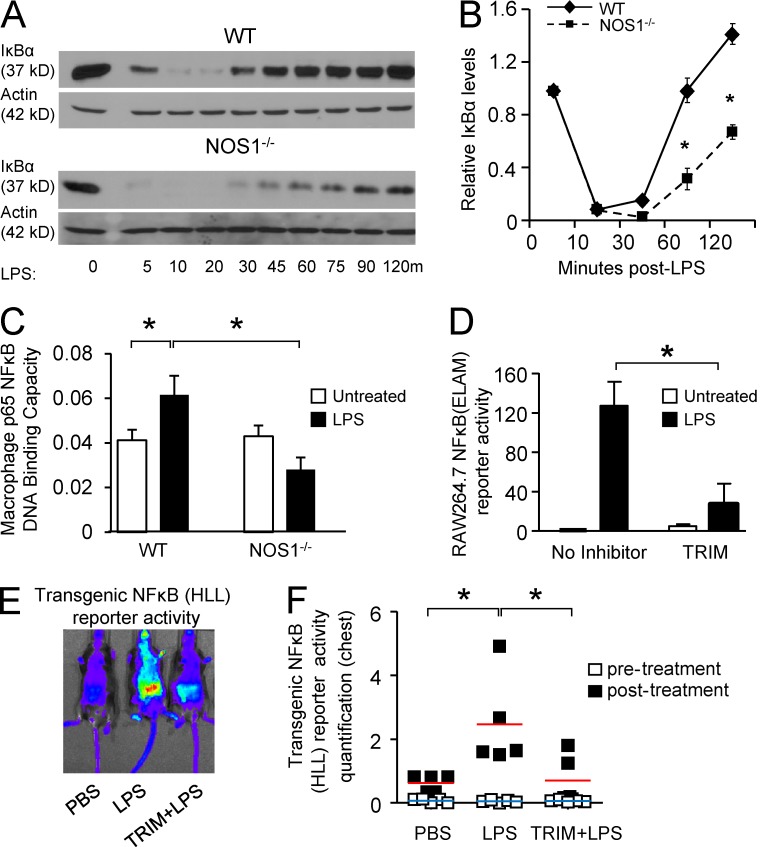
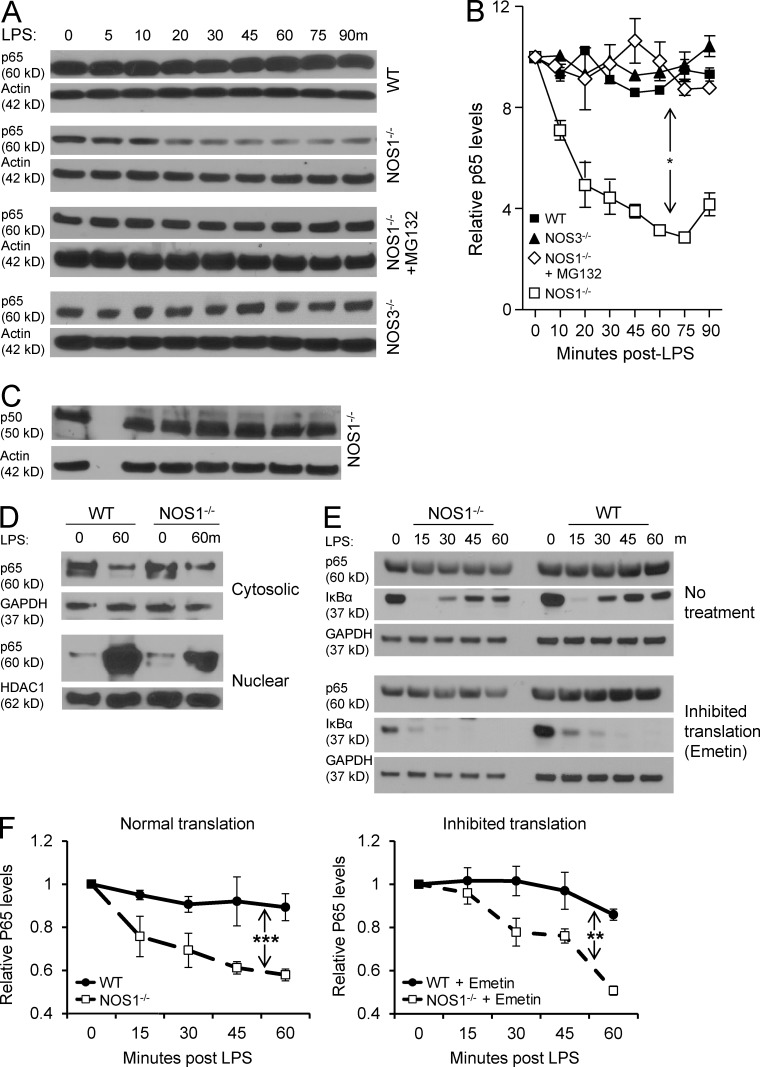
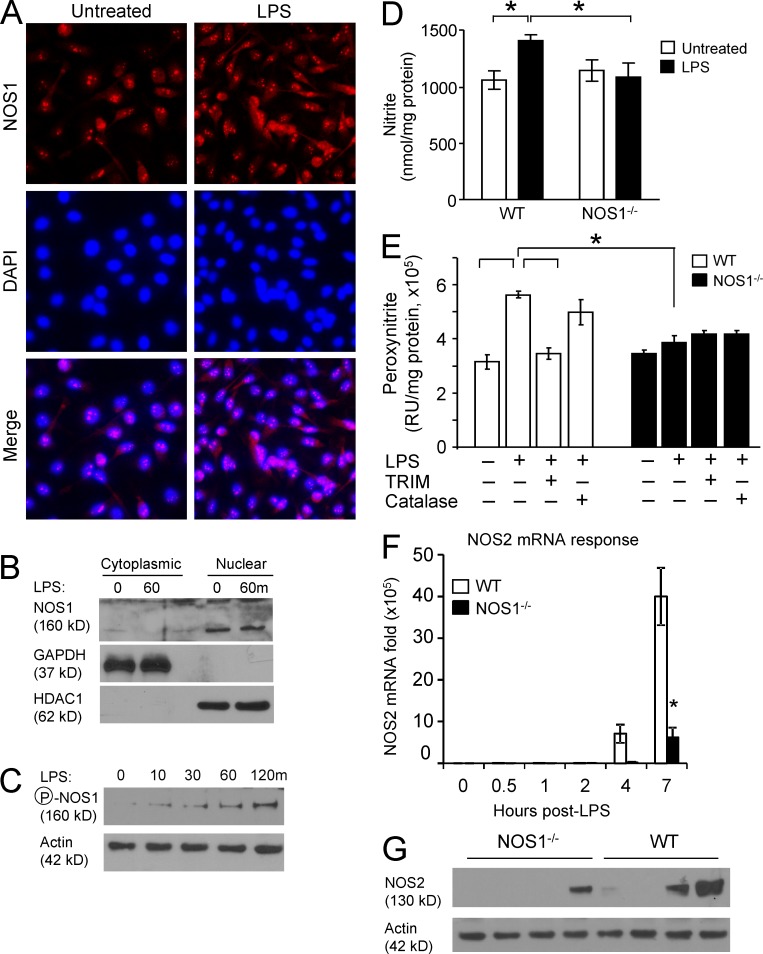
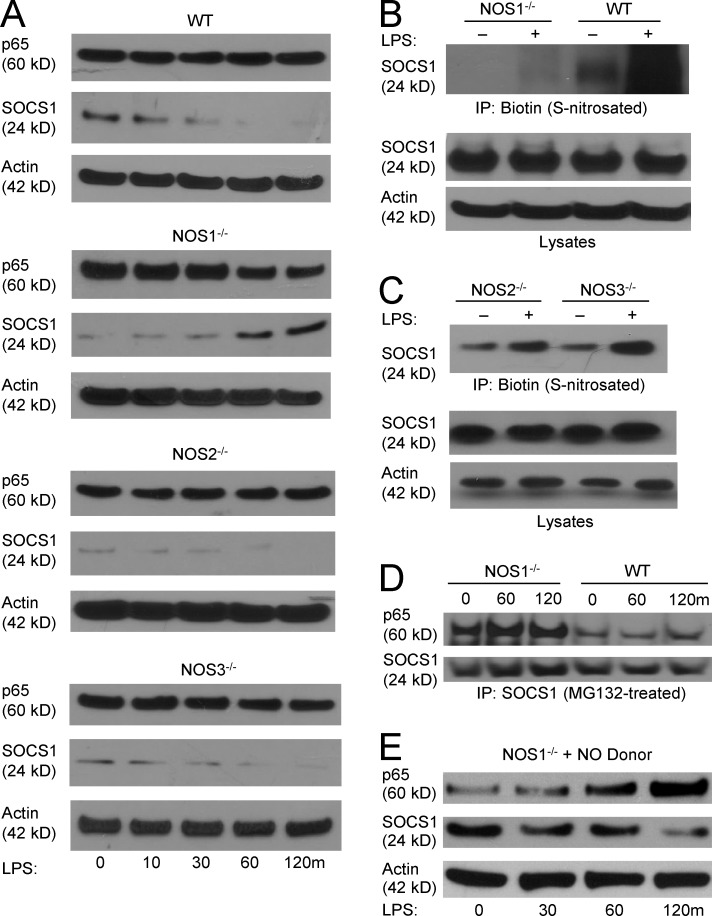
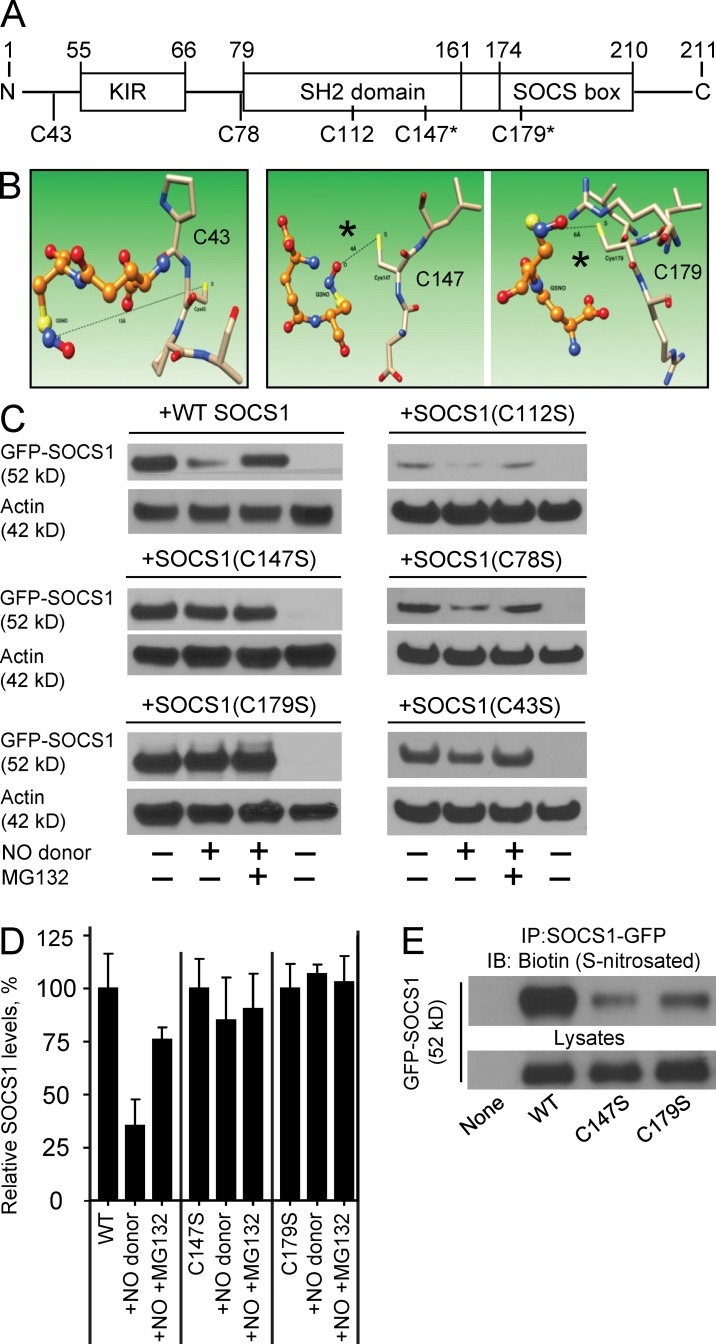
The NF-κB pathway is central to the regulation of inflammation. Here, we demonstrate that the low-output nitric oxide (NO) synthase 1 (NOS1 or nNOS) plays a critical role in the inflammatory response by promoting the activity of NF-κB. Specifically, NOS1-derived NO production in macrophages leads to proteolysis of suppressor of cytokine signaling 1 (SOCS1), alleviating its repression of NF-κB transcriptional activity. As a result, NOS1(-/-) mice demonstrate reduced cytokine production, lung injury, and mortality when subjected to two different models of sepsis. Isolated NOS1(-/-) macrophages demonstrate similar defects in proinflammatory transcription on challenge with Gram-negative bacterial LPS. Consistently, we found that activated NOS1(-/-) macrophages contain increased SOCS1 protein and decreased levels of p65 protein compared with wild-type cells. NOS1-dependent S-nitrosation of SOCS1 impairs its binding to p65 and targets SOCS1 for proteolysis. Treatment of NOS1(-/-) cells with exogenous NO rescues both SOCS1 degradation and stabilization of p65 protein. Point mutation analysis demonstrated that both Cys147 and Cys179 on SOCS1 are required for its NO-dependent degradation. These findings demonstrate a fundamental role for NOS1-derived NO in regulating TLR4-mediated inflammatory gene transcription, as well as the intensity and duration of the resulting host immune response.
© 2015 Baig et al.
Figures
References
-
- Alcamo E., Mizgerd J.P., Horwitz B.H., Bronson R., Beg A.A., Scott M., Doerschuk C.M., Hynes R.O., and Baltimore D.. 2001. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-kappa B in leukocyte recruitment. J. Immunol. 167:1592–1600. 10.4049/jimmunol.167.3.1592 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
