

Evidence for an induced conformational change in the catalytic mechanism of homoisocitrate dehydrogenase for Saccharomyces cerevisiae: Characterization of the D271N mutant enzyme
- PMID: 26325079
- PMCID: PMC4587348
- DOI: 10.1016/j.abb.2015.08.016
Evidence for an induced conformational change in the catalytic mechanism of homoisocitrate dehydrogenase for Saccharomyces cerevisiae: Characterization of the D271N mutant enzyme
Abstract
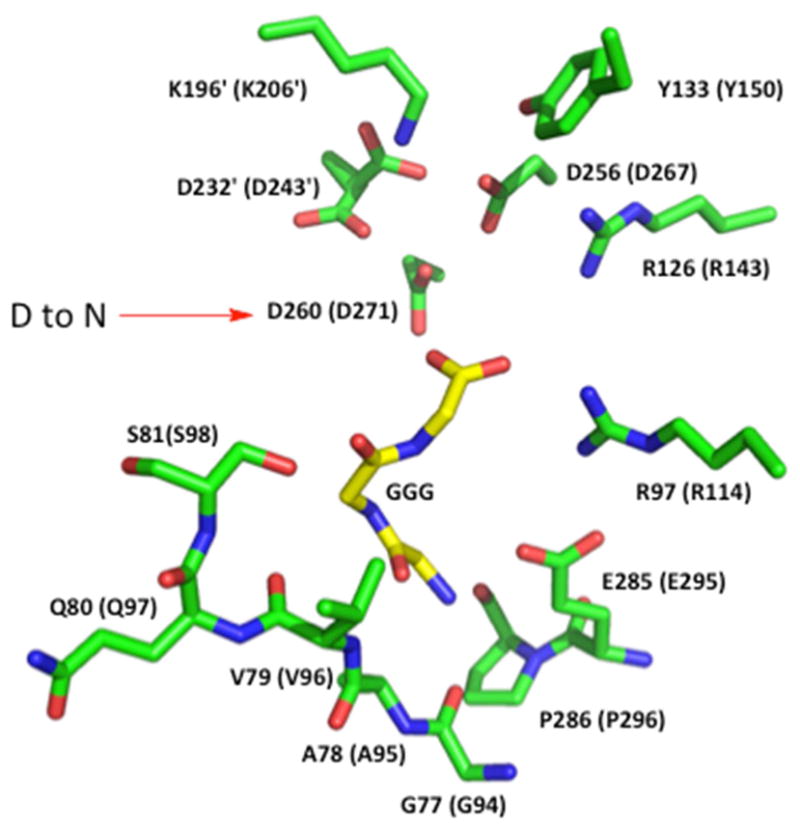
Homoisocitrate dehydrogenase (HIcDH) catalyzes the NAD(+)-dependent oxidative decarboxylation of HIc to α-ketoadipate, the fourth step in the α-aminoadipate pathway responsible for the de novo synthesis of l-lysine in fungi. A mechanism has been proposed for the enzyme that makes use of a Lys-Tyr pair as acid-base catalysts, with Lys acting as a base to accept a proton from the α-hydroxyl of homoisocitrate, and Tyr acting as an acid to protonate the C3 of the enol of α-ketoadipate in the enolization reaction. Three conserved aspartate residues, D243, D267 and D271, coordinate Mg(2+), which is also coordinated to the α-carboxylate and α-hydroxyl of homoisocitrate. On the basis of kinetic isotope effects, it was proposed that a conformational change to close the active site and organize the active site for catalysis contributed to rate limitation of the overall reaction of the Saccharomyces cerevisiae HIcDH (Lin, Y., Volkman, J., Nicholas, K. M., Yamamoto, T., Eguchi, T., Nimmo, S. L., West, A. H., and Cook, P. F. (2008) Biochemistry47, 4169-4180.). In order to test this hypothesis, site-directed mutagenesis was used to change D271, a metal ion ligand and binding determinant for MgHIc, to N. The mutant enzyme was characterized using initial rate studies. A decrease of 520-fold was observed in V and V/KMgHIc, suggesting the same step(s) limit the reaction at limiting and saturating MgHIc concentrations. Solvent kinetic deuterium isotope effects (SKIE) and viscosity effects are consistent with a rate-limiting pre-catalytic conformational change at saturating reactant concentrations. In addition, at limiting MgHIc, an inverse (SKIE) of 0.7 coupled to a significant normal effect of viscosogen (2.1) indicates equilibrium binding of MgHIc prior to the rate-limiting conformational change. The maximum rate exhibits a small partial change at high pH suggesting a pH-dependent conformational change, while V/KMgHIc exhibits the same partial change observed in V, and a decrease at low pH with a pKa of 6 reflecting the requirement for the unprotonated form of MgHIc to bind to enzyme. However, neither parameter reflects the pH dependence of the chemical reaction. This pH independence of the chemical reaction over the range 5.5-9.5 is consistent with the much slower conformational change that would effectively perturb the observed pK values for catalytic groups to lower and higher pH. In other words, the pH dependence of the chemical reaction will only be observed when chemistry becomes slower than the rate of the conformational change. Data support the hypothesis of the existence of a pre-catalytic conformational change coupled to the binding of MgHIc.
Keywords: Homoisocitrate dehydrogenase; Initial rate studies; Isotope effects; Site-directed mutagenesis; Viscosity; pH-Rate profiles.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures




Similar articles
-
Site-directed mutagenesis as a probe of the acid-base catalytic mechanism of homoisocitrate dehydrogenase from Saccharomyces cerevisiae.Biochemistry. 2009 Aug 4;48(30):7305-12. doi: 10.1021/bi900175z. Biochemistry. 2009. PMID: 19530703 Free PMC article.
-
Chemical mechanism of homoisocitrate dehydrogenase from Saccharomyces cerevisiae.Biochemistry. 2008 Apr 1;47(13):4169-80. doi: 10.1021/bi702361j. Epub 2008 Mar 6. Biochemistry. 2008. PMID: 18321070
-
Evidence in support of lysine 77 and histidine 96 as acid-base catalytic residues in saccharopine dehydrogenase from Saccharomyces cerevisiae.Biochemistry. 2012 Jan 31;51(4):857-66. doi: 10.1021/bi201808u. Epub 2012 Jan 23. Biochemistry. 2012. PMID: 22243403 Free PMC article.
-
Perspectives on electrostatics and conformational motions in enzyme catalysis.Acc Chem Res. 2015 Feb 17;48(2):482-9. doi: 10.1021/ar500390e. Epub 2015 Jan 7. Acc Chem Res. 2015. PMID: 25565178 Free PMC article. Review.
-
Probes of mechanism and transition-state structure in the alcohol dehydrogenase reaction.CRC Crit Rev Biochem. 1981;10(1):39-78. doi: 10.3109/10409238109114635. CRC Crit Rev Biochem. 1981. PMID: 7011676 Review.
References
-
- Xu H, Andi B, Qian J, West AH, Cook PF. The α-aminoadipate pathway for lysine biosynthesis in fungi. Cell Biochemistry and Biophysics. 2006;45:43–64. - PubMed
-
- Karsten WE, Cook PF. Pyridine nucleotide dependent α-hydroxyacid oxidative decarboxylases: An overview. Protein Pept Lett. 2000;7:281–286.
-
- Dyson JED, Dorazio RE, Hanson WH. Sheep liver 6-phosphogluconate dehydrogenase: Isolation procedure and effect of pH, ionic-strength, and metal-ions on kinetic parameters. Arch Biochem Biophys. 1973;154:623–635. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
