

An Integrated Framework Advancing Membrane Protein Modeling and Design
- PMID: 26325167
- PMCID: PMC4556676
- DOI: 10.1371/journal.pcbi.1004398
An Integrated Framework Advancing Membrane Protein Modeling and Design
Abstract
Membrane proteins are critical functional molecules in the human body, constituting more than 30% of open reading frames in the human genome. Unfortunately, a myriad of difficulties in overexpression and reconstitution into membrane mimetics severely limit our ability to determine their structures. Computational tools are therefore instrumental to membrane protein structure prediction, consequently increasing our understanding of membrane protein function and their role in disease. Here, we describe a general framework facilitating membrane protein modeling and design that combines the scientific principles for membrane protein modeling with the flexible software architecture of Rosetta3. This new framework, called RosettaMP, provides a general membrane representation that interfaces with scoring, conformational sampling, and mutation routines that can be easily combined to create new protocols. To demonstrate the capabilities of this implementation, we developed four proof-of-concept applications for (1) prediction of free energy changes upon mutation; (2) high-resolution structural refinement; (3) protein-protein docking; and (4) assembly of symmetric protein complexes, all in the membrane environment. Preliminary data show that these algorithms can produce meaningful scores and structures. The data also suggest needed improvements to both sampling routines and score functions. Importantly, the applications collectively demonstrate the potential of combining the flexible nature of RosettaMP with the power of Rosetta algorithms to facilitate membrane protein modeling and design.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
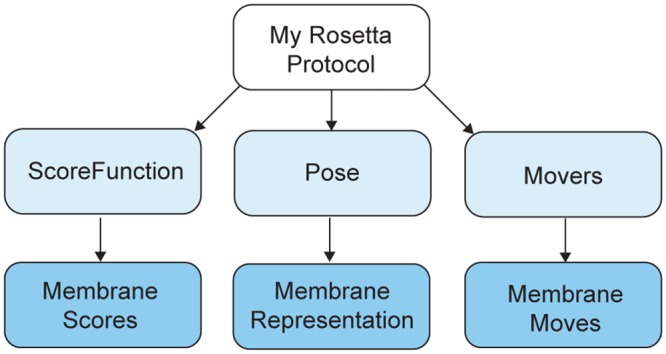
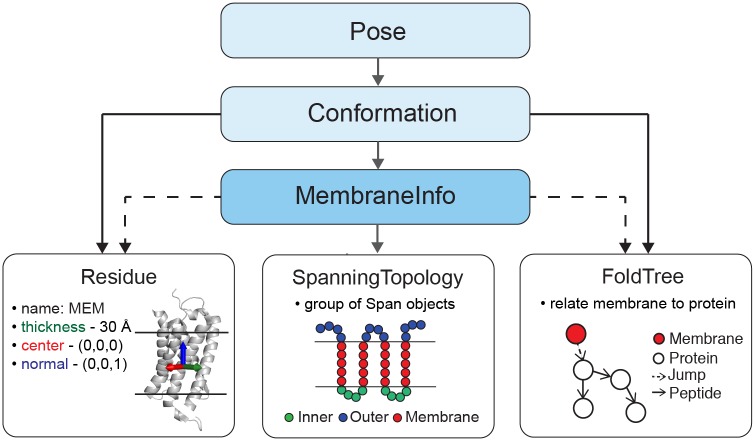

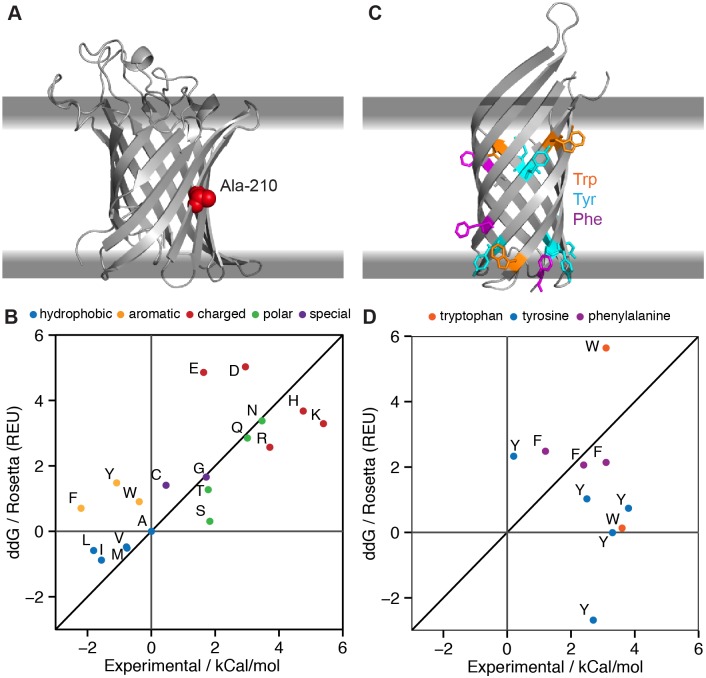
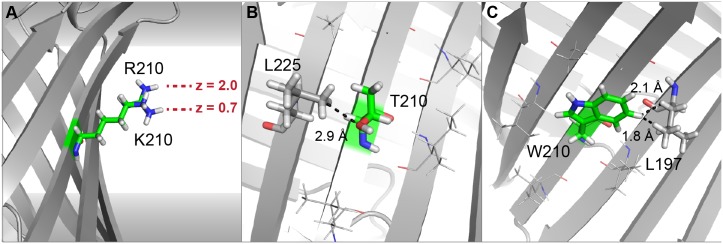
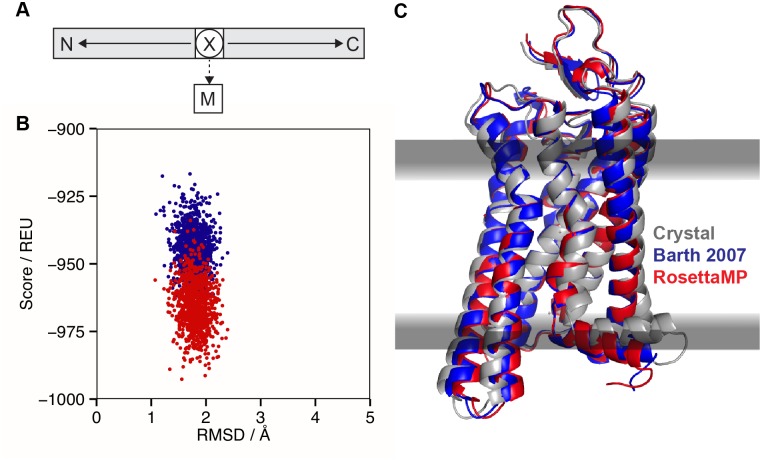
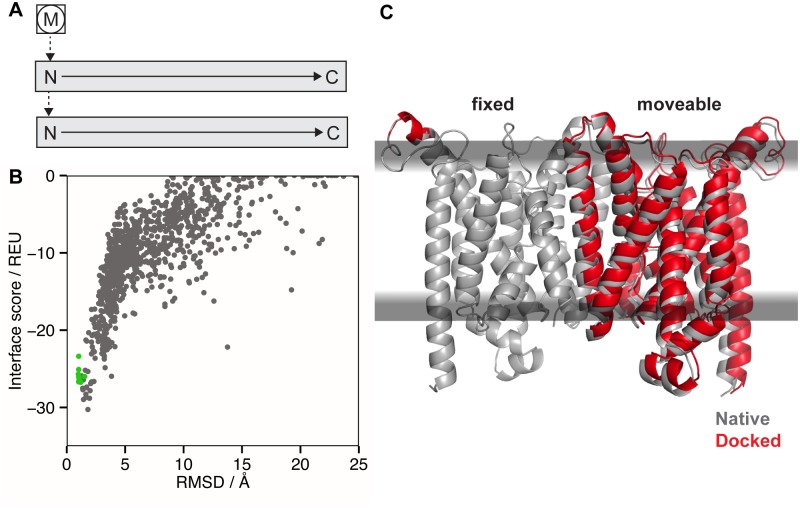
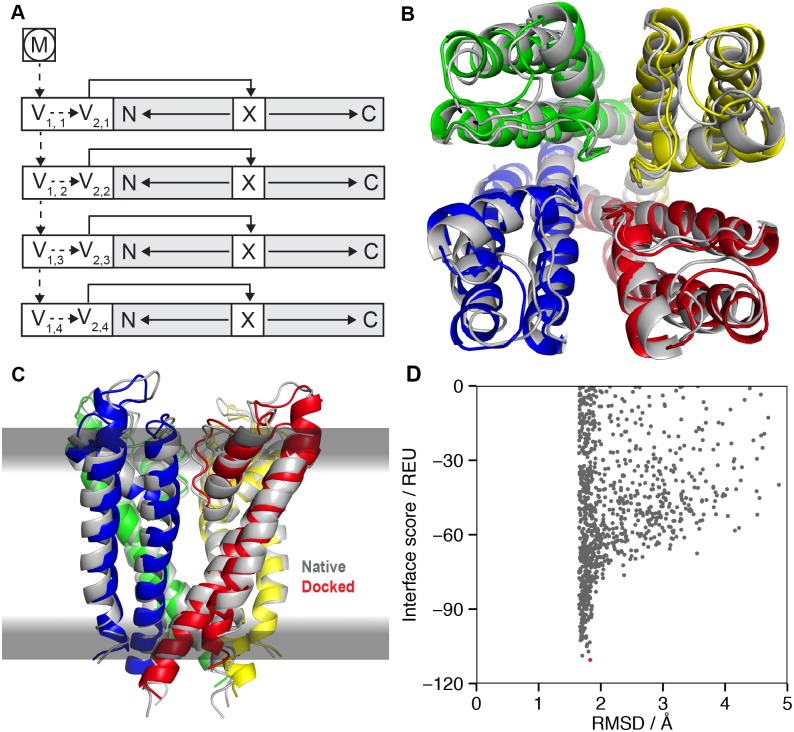
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
