

Endothelial Dysfunction and Amyloid-β-Induced Neurovascular Alterations
- PMID: 26328781
- PMCID: PMC4775455
- DOI: 10.1007/s10571-015-0256-9
Endothelial Dysfunction and Amyloid-β-Induced Neurovascular Alterations
Abstract
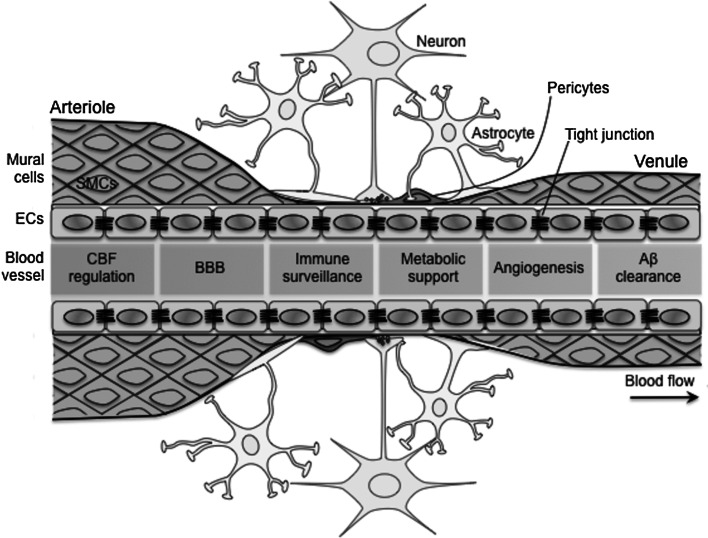
Alzheimer's disease (AD) and cerebrovascular diseases share common vascular risk factors that have disastrous effects on cerebrovascular regulation. Endothelial cells, lining inner walls of cerebral blood vessels, form a dynamic interface between the blood and the brain and are critical for the maintenance of neurovascular homeostasis. Accordingly, injury in endothelial cells is regarded as one of the earliest symptoms of impaired vasoregulatory mechanisms. Extracellular buildup of amyloid-β (Aβ) is a central pathogenic factor in AD. Aβ exerts potent detrimental effects on cerebral blood vessels and impairs endothelial structure and function. Recent evidence implicates vascular oxidative stress and activation of the non-selective cationic channel transient receptor potential melastatin (TRPM)-2 on endothelial cells in the mechanisms of Aβ-induced neurovascular dysfunction. Thus, Aβ triggers opening of TRPM2 channels in endothelial cells leading to intracellular Ca(2+) overload and vasomotor dysfunction. The cerebrovascular dysfunction may contribute to AD pathogenesis by reducing the cerebral blood supply, leading to increased susceptibility to vascular insufficiency, and by promoting Aβ accumulation. The recent realization that vascular factors contribute to AD pathobiology suggests new targets for the prevention and treatment of this devastating disease.
Keywords: Alzheimer’s disease; Cerebral blood flow; Cerebral endothelial cells; TRPM2; β-amyloid.
Figures


References
-
- Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R, Pandya Y, Esh C, Connor DJ, Sabbagh M, Walker DG, Roher AE (2007) Circle of Willis atherosclerosis: association with Alzheimer’s disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol 113(1):13–21. doi:10.1007/s00401-006-0136-y - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
