

Genome-wide replication landscape of Candida glabrata
- PMID: 26329162
- PMCID: PMC4556013
- DOI: 10.1186/s12915-015-0177-6
Genome-wide replication landscape of Candida glabrata
Abstract
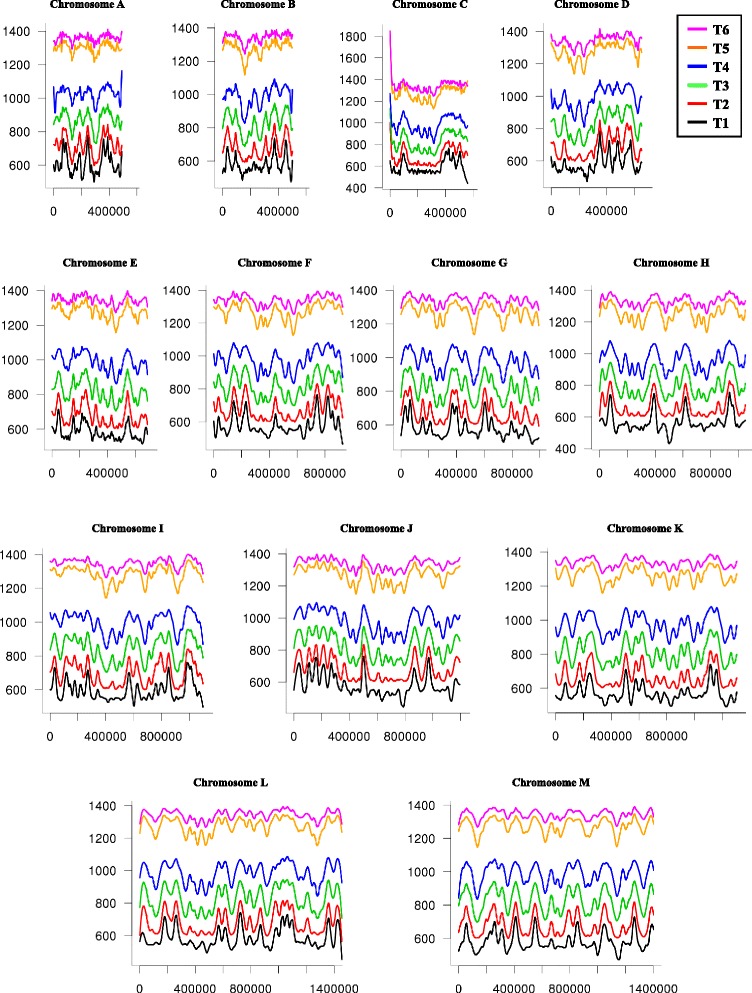
Background: The opportunistic pathogen Candida glabrata is a member of the Saccharomycetaceae yeasts. Like its close relative Saccharomyces cerevisiae, it underwent a whole-genome duplication followed by an extensive loss of genes. Its genome contains a large number of very long tandem repeats, called megasatellites. In order to determine the whole replication program of the C. glabrata genome and its general chromosomal organization, we used deep-sequencing and chromosome conformation capture experiments.
Results: We identified 253 replication fork origins, genome wide. Centromeres, HML and HMR loci, and most histone genes are replicated early, whereas natural chromosomal breakpoints are located in late-replicating regions. In addition, 275 autonomously replicating sequences (ARS) were identified during ARS-capture experiments, and their relative fitness was determined during growth competition. Analysis of ARSs allowed us to identify a 17-bp consensus, similar to the S. cerevisiae ARS consensus sequence but slightly more constrained. Megasatellites are not in close proximity to replication origins or termini. Using chromosome conformation capture, we also show that early origins tend to cluster whereas non-subtelomeric megasatellites do not cluster in the yeast nucleus.
Conclusions: Despite a shorter cell cycle, the C. glabrata replication program shares unexpected striking similarities to S. cerevisiae, in spite of their large evolutionary distance and the presence of highly repetitive large tandem repeats in C. glabrata. No correlation could be found between the replication program and megasatellites, suggesting that their formation and propagation might not be directly caused by replication fork initiation or termination.
Figures






References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
