

Pancreatic Cancer Cell Migration and Metastasis Is Regulated by Chemokine-Biased Agonism and Bioenergetic Signaling
- PMID: 26330165
- PMCID: PMC4560104
- DOI: 10.1158/0008-5472.CAN-14-2645
Pancreatic Cancer Cell Migration and Metastasis Is Regulated by Chemokine-Biased Agonism and Bioenergetic Signaling
Abstract
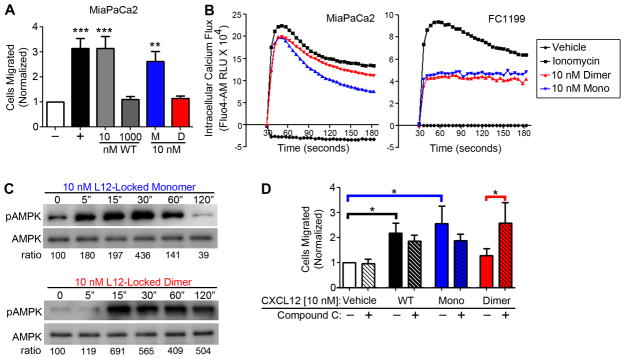
Patients with pancreatic ductal adenocarcinoma (PDAC) invariably succumb to metastatic disease, but the underlying mechanisms that regulate PDAC cell movement and metastasis remain little understood. In this study, we investigated the effects of the chemokine gene CXCL12, which is silenced in PDAC tumors, yet is sufficient to suppress growth and metastasis when re-expressed. Chemokines like CXCL12 regulate cell movement in a biphasic pattern, with peak migration typically in the low nanomolar concentration range. Herein, we tested the hypothesis that the biphasic cell migration pattern induced by CXCL12 reflected a biased agonist bioenergetic signaling that might be exploited to interfere with PDAC metastasis. In human and murine PDAC cell models, we observed that nonmigratory doses of CXCL12 were sufficient to decrease oxidative phosphorylation and glycolytic capacity and to increase levels of phosphorylated forms of the master metabolic kinase AMPK. Those same doses of CXCL12 locked myosin light chain into a phosphorylated state, thereby decreasing F-actin polymerization and preventing cell migration in a manner dependent upon AMPK and the calcium-dependent kinase CAMKII. Notably, at elevated concentrations of CXCL12 that were insufficient to trigger chemotaxis of PDAC cells, AMPK blockade resulted in increased cell movement. In two preclinical mouse models of PDAC, administration of CXCL12 decreased tumor dissemination, supporting our hypothesis that chemokine-biased agonist signaling may offer a useful therapeutic strategy. Our results offer a mechanistic rationale for further investigation of CXCL12 as a potential therapy to prevent or treat PDAC metastasis.
©2015 American Association for Cancer Research.
Conflict of interest statement
Figures






References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: A Cancer Journal for Clinicians. 2012;62(1):10–29. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
