

Stationary-Phase Persisters to Ofloxacin Sustain DNA Damage and Require Repair Systems Only during Recovery
- PMID: 26330511
- PMCID: PMC4556807
- DOI: 10.1128/mBio.00731-15
Stationary-Phase Persisters to Ofloxacin Sustain DNA Damage and Require Repair Systems Only during Recovery
Abstract
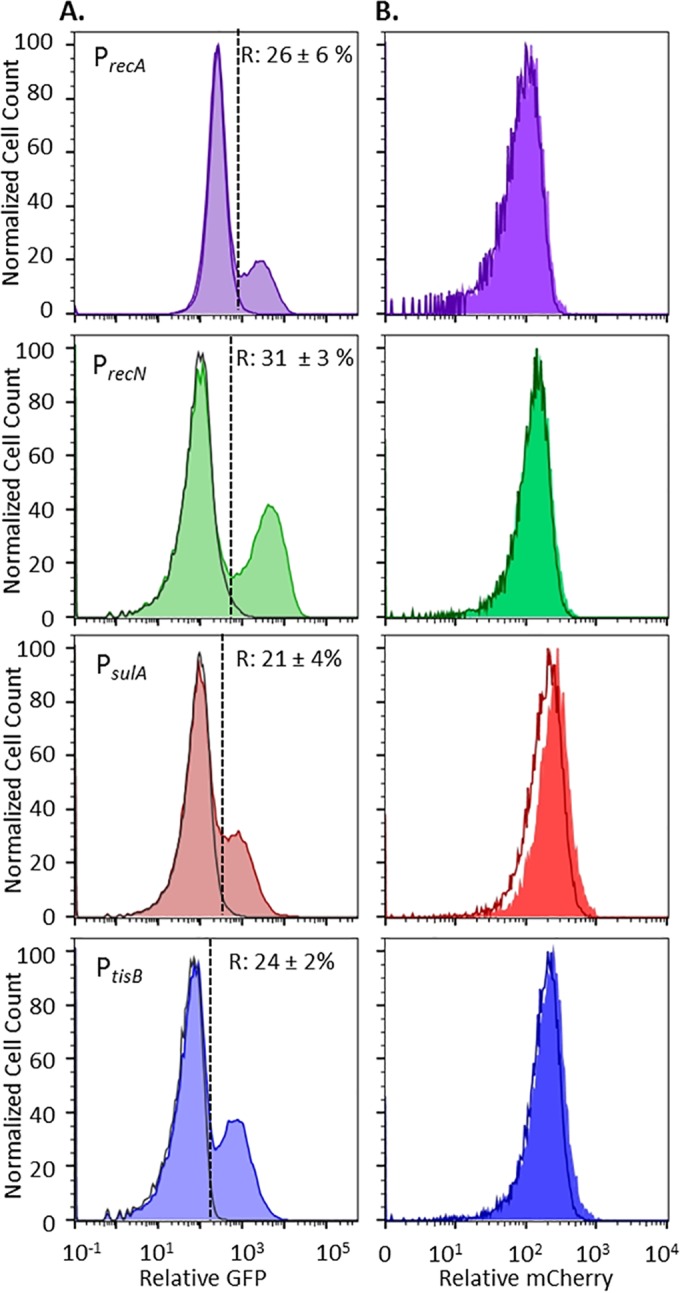
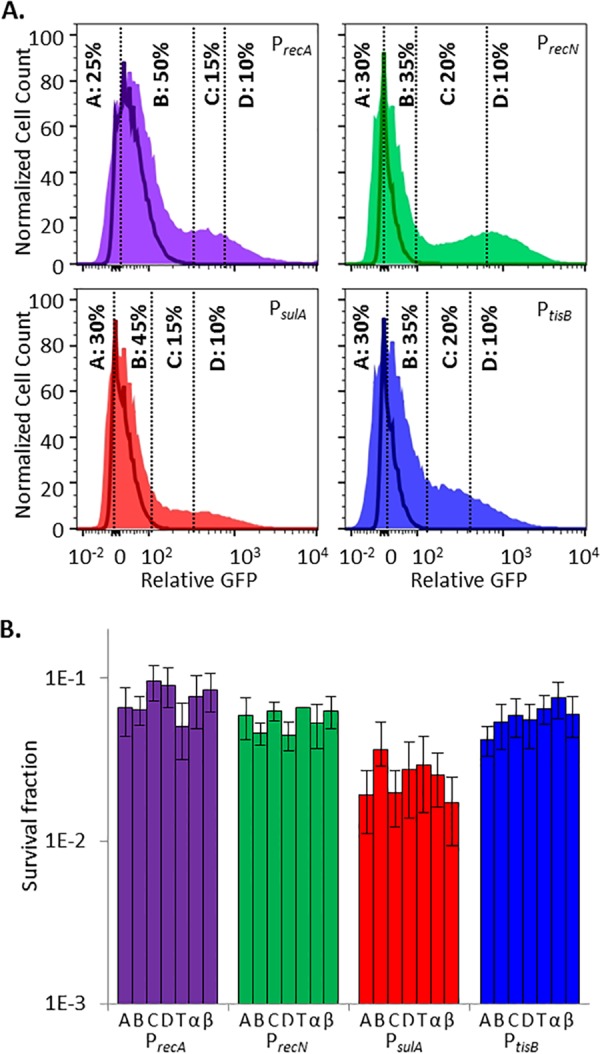
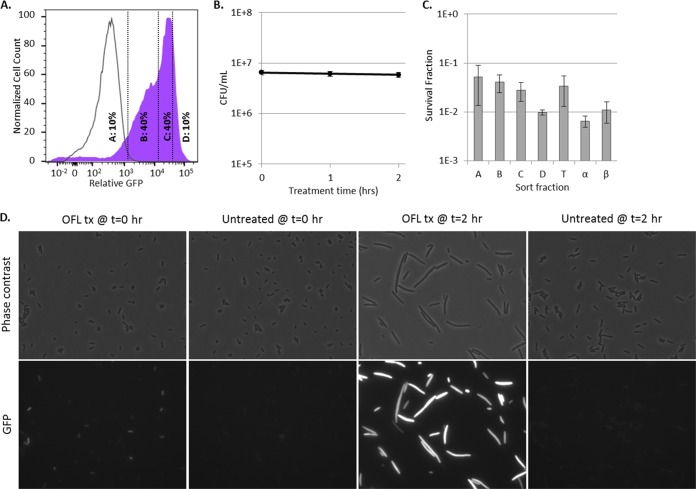
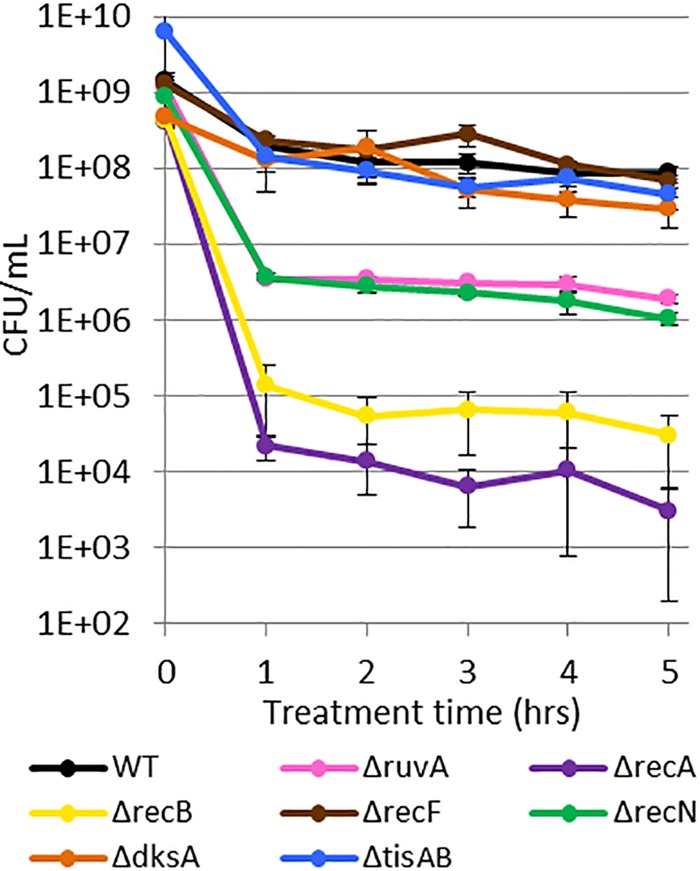
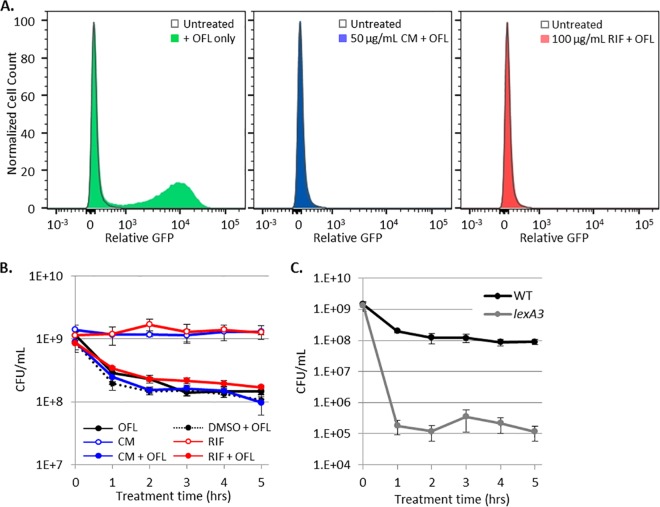
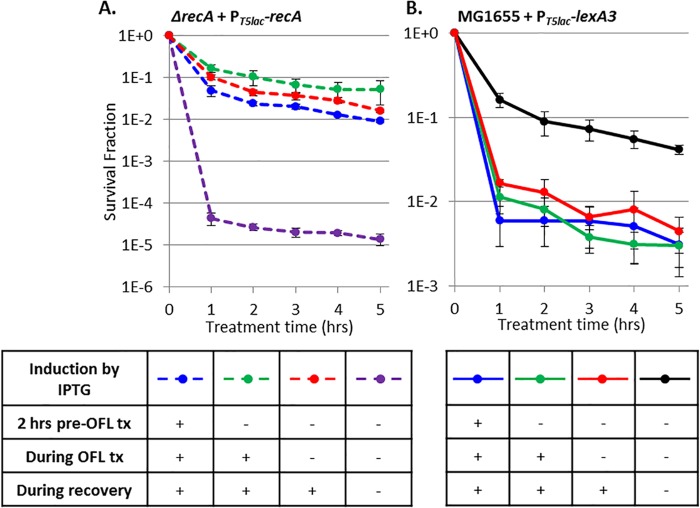
Chronic infections are a serious health care problem, and bacterial persisters have been implicated in infection reoccurrence. Progress toward finding antipersister therapies has been slow, in part because of knowledge gaps regarding the physiology of these rare phenotypic variants. Evidence shows that growth status is important for survival, as nongrowing cultures can have 100-fold more persisters than growing populations. However, additional factors are clearly important, as persisters remain rare even in nongrowing populations. What features, beyond growth inhibition, allow persisters to survive antibiotic stress while the majority of their kin succumb to it remains an open question. To investigate this, we used stationary phase as a model nongrowing environment to study Escherichia coli persistence to ofloxacin. Given that the prevailing model of persistence attributes survival to transient dormancy and antibiotic target inactivity, we anticipated that persisters would suffer less damage than their dying kin. However, using genetic mutants, flow cytometry, fluorescence-activated cell sorting, and persistence assays, we discovered that nongrowing ofloxacin persisters experience antibiotic-induced damage that is indistinguishable from that of nonpersisters. Consistent with this, we found that these persisters required DNA repair for survival and that repair machinery was unnecessary until the posttreatment recovery period (after ofloxacin removal). These findings suggest that persistence to ofloxacin is not engendered solely by reduced antibiotic target corruption, demonstrate that what happens following antibiotic stress can be critical to the persistence phenotype, and support the notion that inhibition of DNA damage repair systems could be an effective strategy to eliminate fluoroquinolone persisters.
Importance: In the absence of resistant mutants, infection reoccurrences can still occur because of persisters, rare bacterial cells that survive antibiotic treatments to repopulate infection sites. Persister survival is attributed to a transient state of dormancy in which a cell's growth and metabolism are significantly reduced and many essential processes are thought to be inactive. Thus, dormancy is believed to protect persisters from antibiotic-induced damage and death. In this work, we show that in nongrowing populations, persisters to ofloxacin experience the same level of antibiotic-induced damage as cells that succumb to the treatment and that their survival critically depends on repair of this damage after the conclusion of treatment. These findings reveal that persistence to ofloxacin is not engendered solely by reduced antibiotic target corruption and highlight that processes following antibiotic stress are important to survival. We hypothesize that effective antipersister therapies may be developed on the basis of this knowledge.
Copyright © 2015 Völzing and Brynildsen.
Figures
Similar articles
-
Timing of DNA damage responses impacts persistence to fluoroquinolones.Proc Natl Acad Sci U S A. 2018 Jul 3;115(27):E6301-E6309. doi: 10.1073/pnas.1804218115. Epub 2018 Jun 18. Proc Natl Acad Sci U S A. 2018. PMID: 29915065 Free PMC article.
-
(p)ppGpp-Dependent Persisters Increase the Fitness of Escherichia coli Bacteria Deficient in Isoaspartyl Protein Repair.Appl Environ Microbiol. 2016 Aug 15;82(17):5444-54. doi: 10.1128/AEM.00623-16. Print 2016 Sep 1. Appl Environ Microbiol. 2016. PMID: 27371580 Free PMC article.
-
Toxin Induction or Inhibition of Transcription or Translation Posttreatment Increases Persistence to Fluoroquinolones.mBio. 2021 Aug 31;12(4):e0198321. doi: 10.1128/mBio.01983-21. Epub 2021 Aug 17. mBio. 2021. PMID: 34399616 Free PMC article.
-
Multidrug tolerance of biofilms and persister cells.Curr Top Microbiol Immunol. 2008;322:107-31. doi: 10.1007/978-3-540-75418-3_6. Curr Top Microbiol Immunol. 2008. PMID: 18453274 Review.
-
Persister cells and the riddle of biofilm survival.Biochemistry (Mosc). 2005 Feb;70(2):267-74. doi: 10.1007/s10541-005-0111-6. Biochemistry (Mosc). 2005. PMID: 15807669 Review.
Cited by
-
Intoxication of antibiotic persisters by host RNS inactivates their efflux machinery during infection.PLoS Pathog. 2024 Feb 29;20(2):e1012033. doi: 10.1371/journal.ppat.1012033. eCollection 2024 Feb. PLoS Pathog. 2024. PMID: 38421944 Free PMC article.
-
Preexisting variation in DNA damage response predicts the fate of single mycobacteria under stress.EMBO J. 2019 Nov 15;38(22):e101876. doi: 10.15252/embj.2019101876. Epub 2019 Oct 4. EMBO J. 2019. PMID: 31583725 Free PMC article.
-
Toxins targeting transfer RNAs: Translation inhibition by bacterial toxin-antitoxin systems.Wiley Interdiscip Rev RNA. 2019 Jan;10(1):e1506. doi: 10.1002/wrna.1506. Epub 2018 Sep 16. Wiley Interdiscip Rev RNA. 2019. PMID: 30296016 Free PMC article. Review.
-
Cellular Self-Digestion and Persistence in Bacteria.Microorganisms. 2021 Oct 31;9(11):2269. doi: 10.3390/microorganisms9112269. Microorganisms. 2021. PMID: 34835393 Free PMC article. Review.
-
Investigating the Effects of Osmolytes and Environmental pH on Bacterial Persisters.Antimicrob Agents Chemother. 2020 Apr 21;64(5):e02393-19. doi: 10.1128/AAC.02393-19. Print 2020 Apr 21. Antimicrob Agents Chemother. 2020. PMID: 32094133 Free PMC article.
References
-
- Scott RD., II 2009. The direct medical costs of health care-associated infections in U.S. hospitals and the benefits of prevention. Centers for Disease Control and Prevention, Atlanta, GA: http://www.cdc.gov/HAI/pdfs/hai/Scott_CostPaper.pdf.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
