

Trichostatin A Modulates Angiotensin II-induced Vasoconstriction and Blood Pressure Via Inhibition of p66shc Activation
- PMID: 26330760
- PMCID: PMC4553407
- DOI: 10.4196/kjpp.2015.19.5.467
Trichostatin A Modulates Angiotensin II-induced Vasoconstriction and Blood Pressure Via Inhibition of p66shc Activation
Abstract
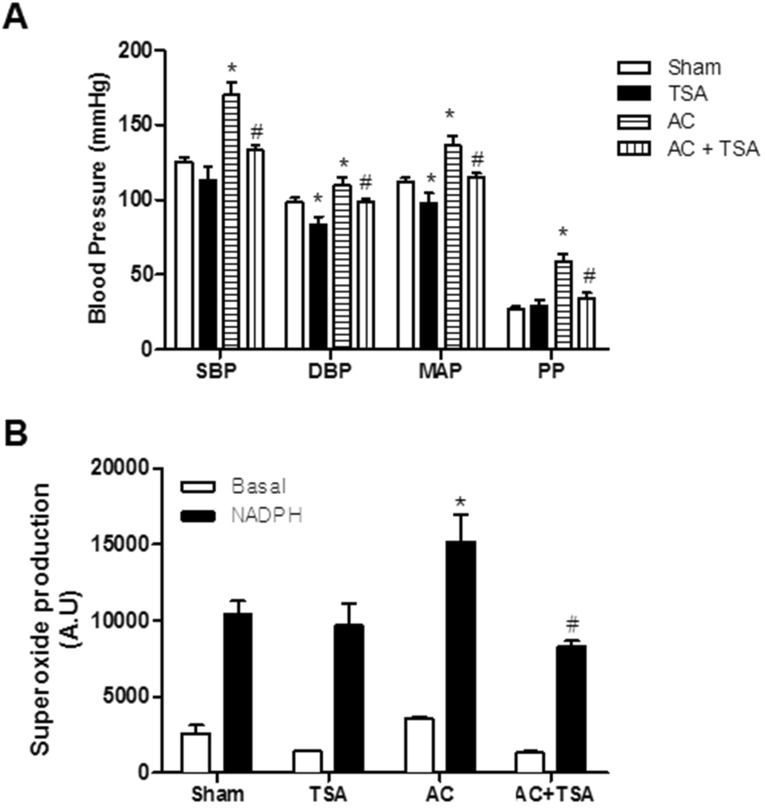
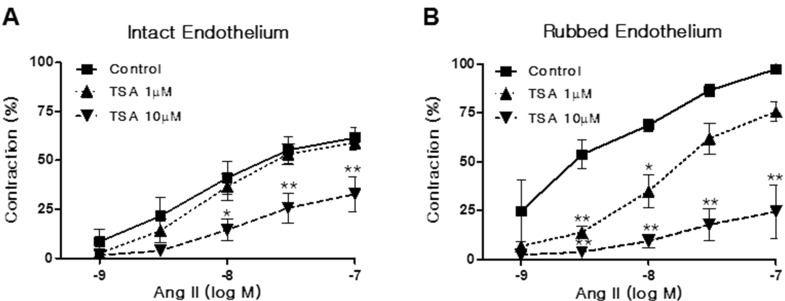
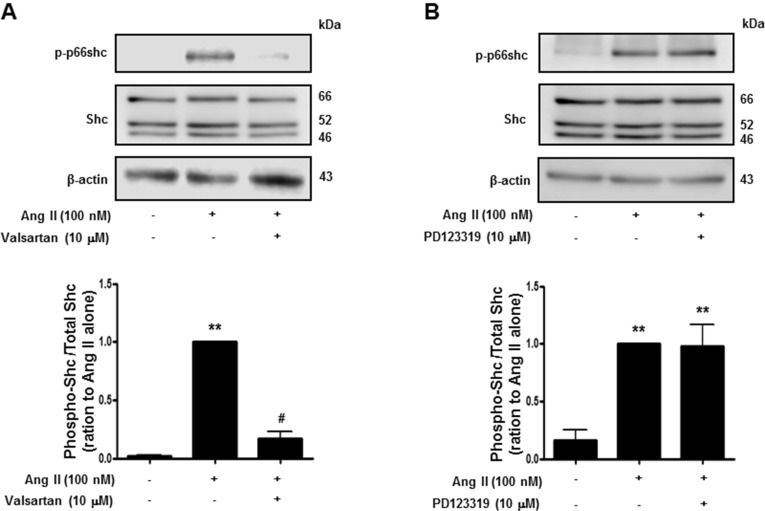
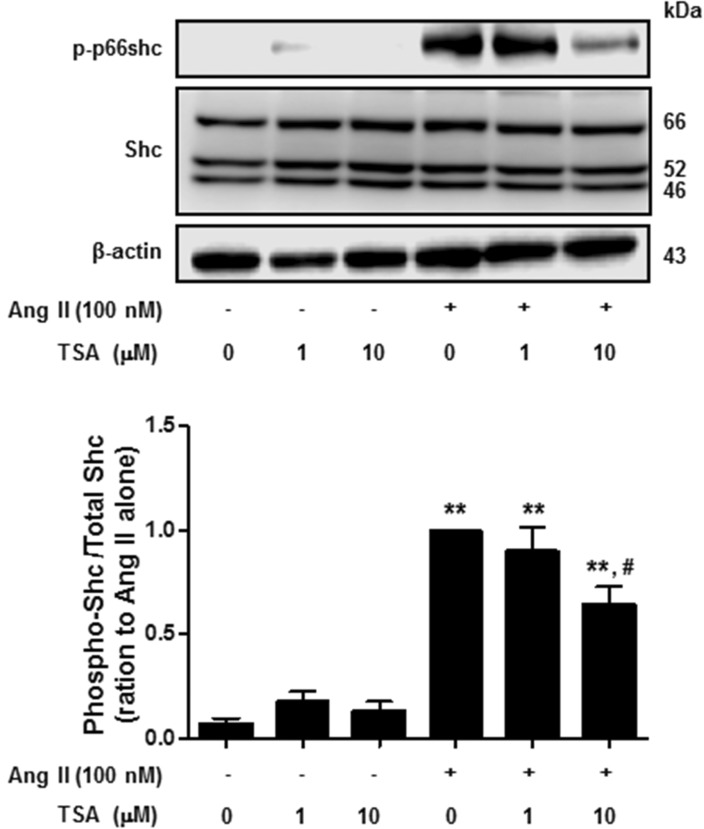
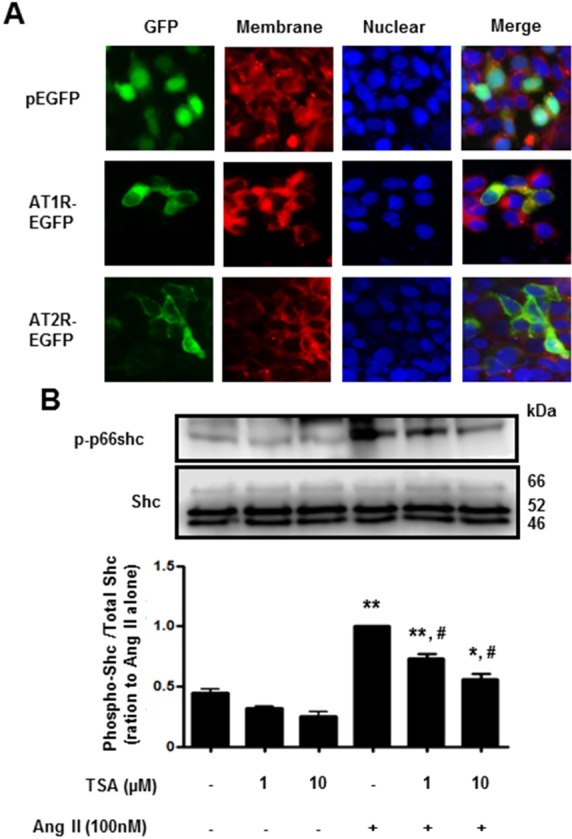
Histone deacetylase (HDAC) has been recognized as a potentially useful therapeutic target for cardiovascular disorders. However, the effect of the HDAC inhibitor, trichostatin A (TSA), on vasoreactivity and hypertension remains unknown. We performed aortic coarctation at the inter-renal level in rats in order to create a hypertensive rat model. Hypertension induced by abdominal aortic coarctation was significantly suppressed by chronic treatment with TSA (0.5 mg/kg/day for 7 days). Nicotinamide adenine dinucleotide phosphate-driven reactive oxygen species production was also reduced in the aortas of TSA-treated aortic coarctation rats. The vasoconstriction induced by angiotensin II (Ang II, 100 nM) was inhibited by TSA in both endothelium-intact and endothelium-denuded rat aortas, suggesting that TSA has mainly acted in vascular smooth muscle cells (VSMCs). In cultured rat aortic VSMCs, Ang II increased p66shc phosphorylation, which was inhibited by the Ang II receptor type I (AT1R) inhibitor, valsartan (10 µM), but not by the AT2R inhibitor, PD123319. TSA (1~10 µM) inhibited Ang II-induced p66shc phosphorylation in VSMCs and in HEK293T cells expressing AT1R. Taken together, these results suggest that TSA treatment inhibited vasoconstriction and hypertension via inhibition of Ang II-induced phosphorylation of p66shc through AT1R.
Keywords: Angiotensin II; Angiotensin receptor type I; Hypertension; Trichostatin A; p66shc.
Conflict of interest statement
Figures
Similar articles
-
Class I histone deacetylase inhibitor MS-275 attenuates vasoconstriction and inflammation in angiotensin II-induced hypertension.PLoS One. 2019 Mar 4;14(3):e0213186. doi: 10.1371/journal.pone.0213186. eCollection 2019. PLoS One. 2019. PMID: 30830950 Free PMC article.
-
Negative regulation of RhoA/Rho kinase by angiotensin II type 2 receptor in vascular smooth muscle cells: role in angiotensin II-induced vasodilation in stroke-prone spontaneously hypertensive rats.J Hypertens. 2005 May;23(5):1037-45. doi: 10.1097/01.hjh.0000166845.49850.39. J Hypertens. 2005. PMID: 15834290
-
Angiotensin II type 2 receptors contribute to vascular responses in spontaneously hypertensive rats treated with angiotensin II type 1 receptor antagonists.Am J Hypertens. 2005 Apr;18(4 Pt 1):493-9. doi: 10.1016/j.amjhyper.2004.11.007. Am J Hypertens. 2005. PMID: 15831358
-
Dissociation of angiotensin II-stimulated activation of mitogen-activated protein kinase kinase from vascular contraction.J Pharmacol Exp Ther. 1998 Sep;286(3):1431-8. J Pharmacol Exp Ther. 1998. PMID: 9732408
-
Angiotensin II signaling via type 2 receptors in a human model of vascular hyporeactivity: implications for hypertension.J Hypertens. 2010 Jan;28(1):111-8. doi: 10.1097/HJH.0b013e328332b738. J Hypertens. 2010. PMID: 19797979
Cited by
-
Tributyrin Intake Attenuates Angiotensin II-Induced Abdominal Aortic Aneurysm in LDLR-/- Mice.Int J Mol Sci. 2023 Apr 28;24(9):8008. doi: 10.3390/ijms24098008. Int J Mol Sci. 2023. PMID: 37175712 Free PMC article.
-
Protein kinase C beta II upregulates intercellular adhesion molecule-1 via mitochondrial activation in cultured endothelial cells.Korean J Physiol Pharmacol. 2017 Jul;21(4):377-384. doi: 10.4196/kjpp.2017.21.4.377. Epub 2017 Jun 26. Korean J Physiol Pharmacol. 2017. PMID: 28706451 Free PMC article.
-
Histone Deacetylases (HDACs) and Atherosclerosis: A Mechanistic and Pharmacological Review.Front Cell Dev Biol. 2020 Nov 12;8:581015. doi: 10.3389/fcell.2020.581015. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33282862 Free PMC article. Review.
-
Wogonin attenuates vascular remodeling by inhibiting smooth muscle cell proliferation and migration in hypertensive rat.Korean J Physiol Pharmacol. 2024 Jan 1;28(1):39-48. doi: 10.4196/kjpp.2024.28.1.39. Korean J Physiol Pharmacol. 2024. PMID: 38154963 Free PMC article.
-
Class I histone deacetylase inhibitor MS-275 attenuates vasoconstriction and inflammation in angiotensin II-induced hypertension.PLoS One. 2019 Mar 4;14(3):e0213186. doi: 10.1371/journal.pone.0213186. eCollection 2019. PLoS One. 2019. PMID: 30830950 Free PMC article.
References
-
- Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell. 1997;89:373–380. - PubMed
-
- Usui T, Okada M, Mizuno W, Oda M, Ide N, Morita T, Hara Y, Yamawaki H. HDAC4 mediates development of hypertension via vascular inflammation in spontaneous hypertensive rats. Am J Physiol Heart Circ Physiol. 2012;302:H1894–H1904. - PubMed
-
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. - PubMed
-
- Pellegrini M, Baldari CT. Apoptosis and oxidative stress-related diseases: the p66Shc connection. Curr Mol Med. 2009;9:392–398. - PubMed
-
- Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
