

The spliceosome is a therapeutic vulnerability in MYC-driven cancer
- PMID: 26331541
- PMCID: PMC4831063
- DOI: 10.1038/nature14985
The spliceosome is a therapeutic vulnerability in MYC-driven cancer
Abstract
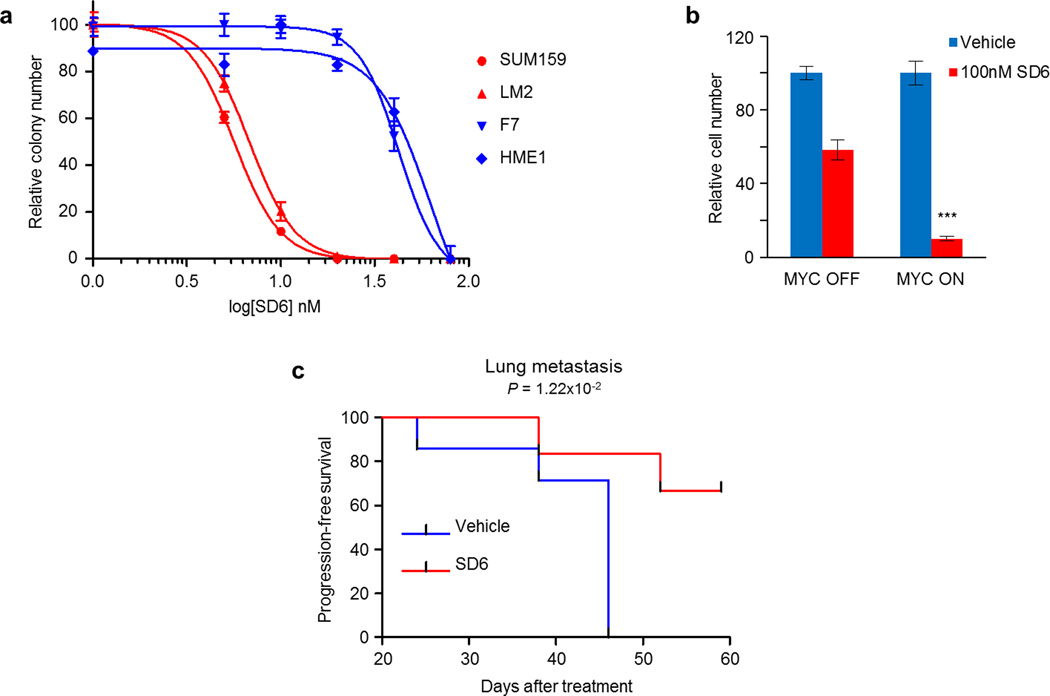
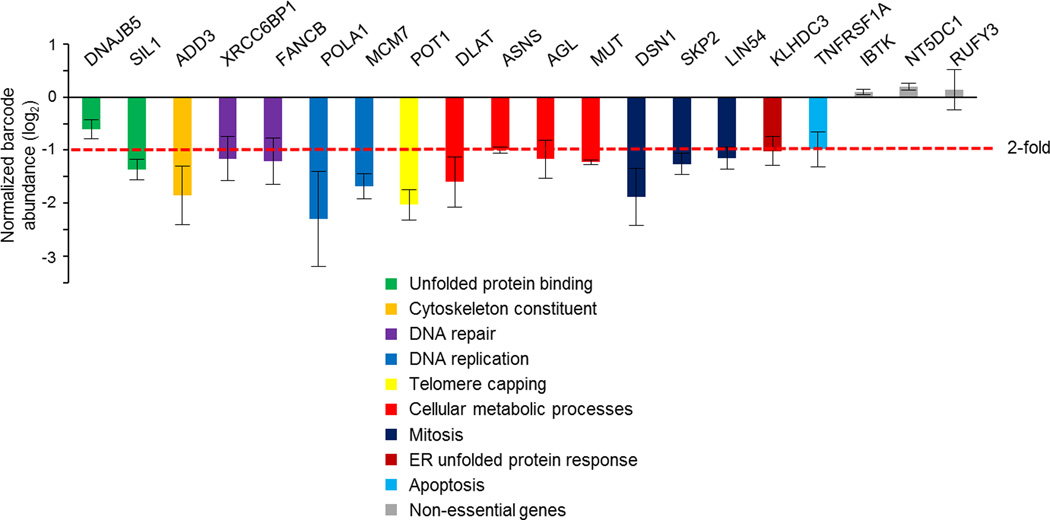
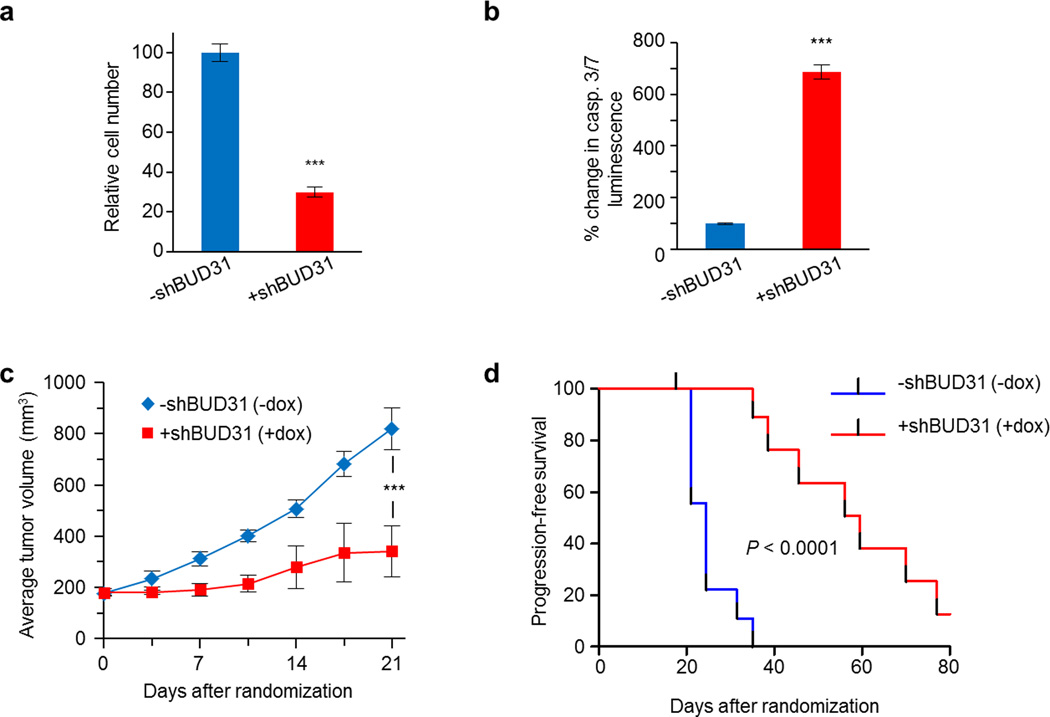
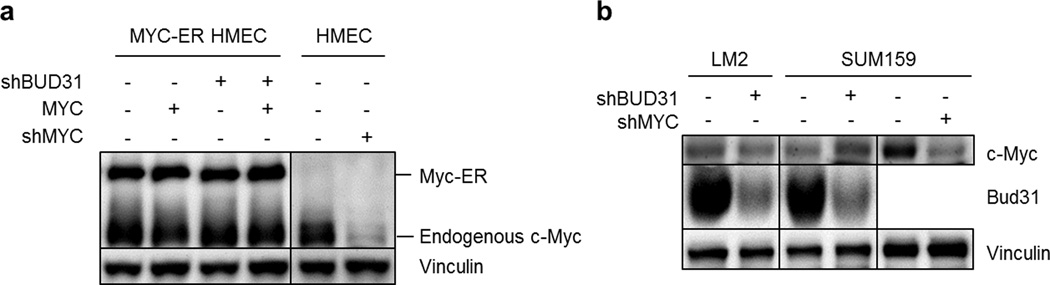
MYC (also known as c-MYC) overexpression or hyperactivation is one of the most common drivers of human cancer. Despite intensive study, the MYC oncogene remains recalcitrant to therapeutic inhibition. MYC is a transcription factor, and many of its pro-tumorigenic functions have been attributed to its ability to regulate gene expression programs. Notably, oncogenic MYC activation has also been shown to increase total RNA and protein production in many tissue and disease contexts. While such increases in RNA and protein production may endow cancer cells with pro-tumour hallmarks, this increase in synthesis may also generate new or heightened burden on MYC-driven cancer cells to process these macromolecules properly. Here we discover that the spliceosome is a new target of oncogenic stress in MYC-driven cancers. We identify BUD31 as a MYC-synthetic lethal gene in human mammary epithelial cells, and demonstrate that BUD31 is a component of the core spliceosome required for its assembly and catalytic activity. Core spliceosomal factors (such as SF3B1 and U2AF1) associated with BUD31 are also required to tolerate oncogenic MYC. Notably, MYC hyperactivation induces an increase in total precursor messenger RNA synthesis, suggesting an increased burden on the core spliceosome to process pre-mRNA. In contrast to normal cells, partial inhibition of the spliceosome in MYC-hyperactivated cells leads to global intron retention, widespread defects in pre-mRNA maturation, and deregulation of many essential cell processes. Notably, genetic or pharmacological inhibition of the spliceosome in vivo impairs survival, tumorigenicity and metastatic proclivity of MYC-dependent breast cancers. Collectively, these data suggest that oncogenic MYC confers a collateral stress on splicing, and that components of the spliceosome may be therapeutic entry points for aggressive MYC-driven cancers.
Figures










Comment in
-
MYC: Splicing up your survival.Nat Rev Cancer. 2015 Oct;15(10):574-5. doi: 10.1038/nrc4013. Epub 2015 Sep 18. Nat Rev Cancer. 2015. PMID: 26383137 No abstract available.
-
Splicing and Dicing MYC-Mediated Synthetic Lethality.Cancer Cell. 2015 Oct 12;28(4):405-406. doi: 10.1016/j.ccell.2015.09.016. Cancer Cell. 2015. PMID: 26461086
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
- R01 CA140474/CA/NCI NIH HHS/United States
- R01 CA178039/CA/NCI NIH HHS/United States
- F30 CA180447/CA/NCI NIH HHS/United States
- R25 GM056929/GM/NIGMS NIH HHS/United States
- P30 AI036211/AI/NIAID NIH HHS/United States
- 1R01CA178039-01/CA/NCI NIH HHS/United States
- S10 RR024574/RR/NCRR NIH HHS/United States
- U54 CA149196/CA/NCI NIH HHS/United States
- R01 GM082837/GM/NIGMS NIH HHS/United States
- 1F30CA180447/CA/NCI NIH HHS/United States
- R01 AR045653/AR/NIAMS NIH HHS/United States
- P30CA125123/CA/NCI NIH HHS/United States
- R01 CA190467/CA/NCI NIH HHS/United States
- U54-CA149196/CA/NCI NIH HHS/United States
- R01 AR060733/AR/NIAMS NIH HHS/United States
- P30 CA125123/CA/NCI NIH HHS/United States
- R21 CA185769/CA/NCI NIH HHS/United States
- R01 HL045565/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
