

Multi-objective optimization for RNA design with multiple target secondary structures
- PMID: 26335276
- PMCID: PMC4559319
- DOI: 10.1186/s12859-015-0706-x
Multi-objective optimization for RNA design with multiple target secondary structures
Abstract
Background: RNAs are attractive molecules as the biological parts for synthetic biology. In particular, the ability of conformational changes, which can be encoded in designer RNAs, enables us to create multistable molecular switches that function in biological circuits. Although various algorithms for designing such RNA switches have been proposed, the previous algorithms optimize the RNA sequences against the weighted sum of objective functions, where empirical weights among objective functions are used. In addition, an RNA design algorithm for multiple pseudoknot targets is currently not available.
Results: We developed a novel computational tool for automatically designing RNA sequences which fold into multiple target secondary structures. Our algorithm designs RNA sequences based on multi-objective genetic algorithm, by which we can explore the RNA sequences having good objective function values without empirical weight parameters among the objective functions. Our algorithm has great flexibility by virtue of this weight-free nature. We benchmarked our multi-target RNA design algorithm with the datasets of two, three, and four target structures and found that our algorithm shows better or comparable design performances compared with the previous algorithms, RNAdesign and Frnakenstein. In addition to the benchmarks with pseudoknot-free datasets, we benchmarked MODENA with two-target pseudoknot datasets and found that MODENA can design the RNAs which have the target pseudoknotted secondary structures whose free energies are close to the lowest free energy. Moreover, we applied our algorithm to a ribozyme-based ON-switch which takes a ribozyme-inactive secondary structure when the theophylline aptamer structure is assumed.
Conclusions: Currently, MODENA is the only RNA design software which can be applied to multiple pseudoknot targets. Successful design results for the multiple targets and an RNA device indicate usefulness of our multi-objective RNA design algorithm.
Figures

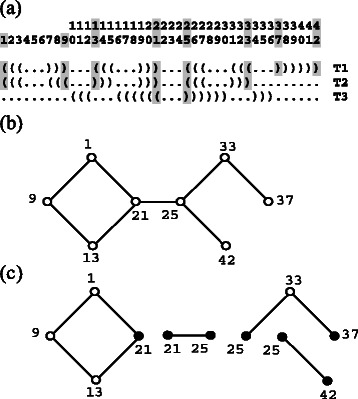
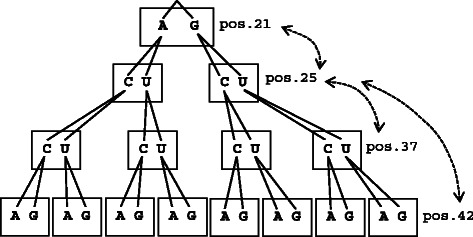




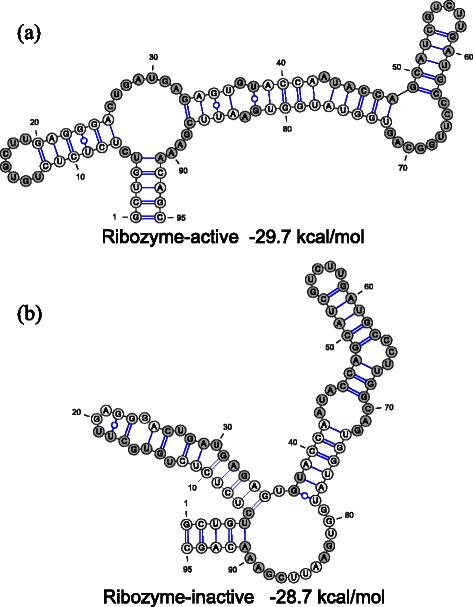
Similar articles
-
A Web Server for Designing Molecular Switches Composed of Two Interacting RNAs.Int J Mol Sci. 2021 Mar 8;22(5):2720. doi: 10.3390/ijms22052720. Int J Mol Sci. 2021. PMID: 33800268 Free PMC article.
-
Multi-objective genetic algorithm for pseudoknotted RNA sequence design.Front Genet. 2012 Apr 26;3:36. doi: 10.3389/fgene.2012.00036. eCollection 2012. Front Genet. 2012. PMID: 22558001 Free PMC article.
-
Computational design of RNAs with complex energy landscapes.Biopolymers. 2013 Dec;99(12):1124-36. doi: 10.1002/bip.22337. Biopolymers. 2013. PMID: 23818234
-
How to benchmark RNA secondary structure prediction accuracy.Methods. 2019 Jun 1;162-163:60-67. doi: 10.1016/j.ymeth.2019.04.003. Epub 2019 Apr 2. Methods. 2019. PMID: 30951834 Free PMC article. Review.
-
Energy-based RNA consensus secondary structure prediction in multiple sequence alignments.Methods Mol Biol. 2014;1097:125-41. doi: 10.1007/978-1-62703-709-9_7. Methods Mol Biol. 2014. PMID: 24639158 Review.
Cited by
-
RNAblueprint: flexible multiple target nucleic acid sequence design.Bioinformatics. 2017 Sep 15;33(18):2850-2858. doi: 10.1093/bioinformatics/btx263. Bioinformatics. 2017. PMID: 28449031 Free PMC article.
-
A Web Server for Designing Molecular Switches Composed of Two Interacting RNAs.Int J Mol Sci. 2021 Mar 8;22(5):2720. doi: 10.3390/ijms22052720. Int J Mol Sci. 2021. PMID: 33800268 Free PMC article.
-
Infrared: a declarative tree decomposition-powered framework for bioinformatics.Algorithms Mol Biol. 2024 Mar 16;19(1):13. doi: 10.1186/s13015-024-00258-2. Algorithms Mol Biol. 2024. PMID: 38493130 Free PMC article.
-
Design Principles of Biological Oscillators through Optimization: Forward and Reverse Analysis.PLoS One. 2016 Dec 15;11(12):e0166867. doi: 10.1371/journal.pone.0166867. eCollection 2016. PLoS One. 2016. PMID: 27977695 Free PMC article.
-
Automated Design of Diverse Stand-Alone Riboswitches.ACS Synth Biol. 2019 Aug 16;8(8):1838-1846. doi: 10.1021/acssynbio.9b00142. Epub 2019 Jul 29. ACS Synth Biol. 2019. PMID: 31298841 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
