

Membrane Repair: Mechanisms and Pathophysiology
- PMID: 26336031
- PMCID: PMC4600952
- DOI: 10.1152/physrev.00037.2014
Membrane Repair: Mechanisms and Pathophysiology
Abstract
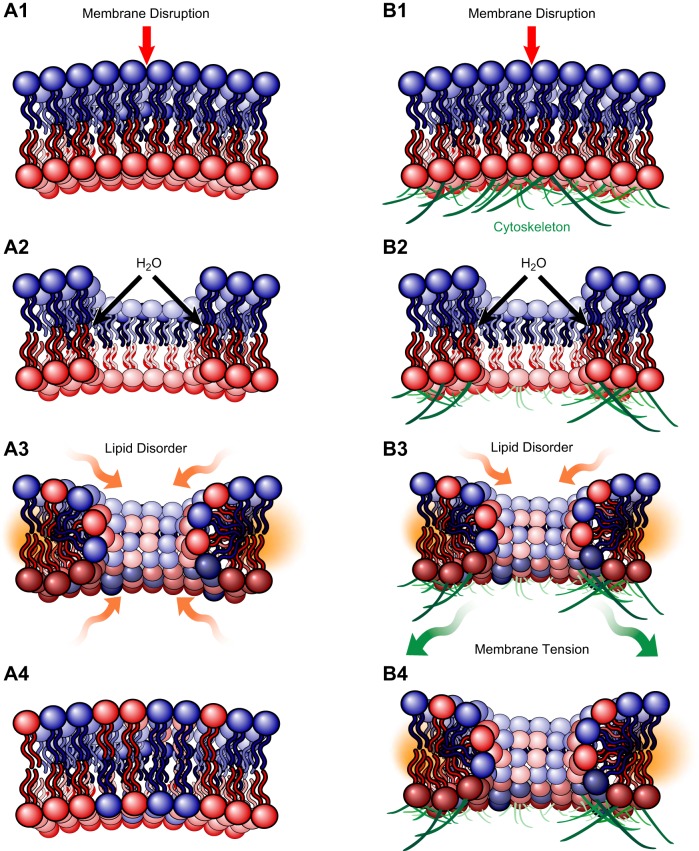
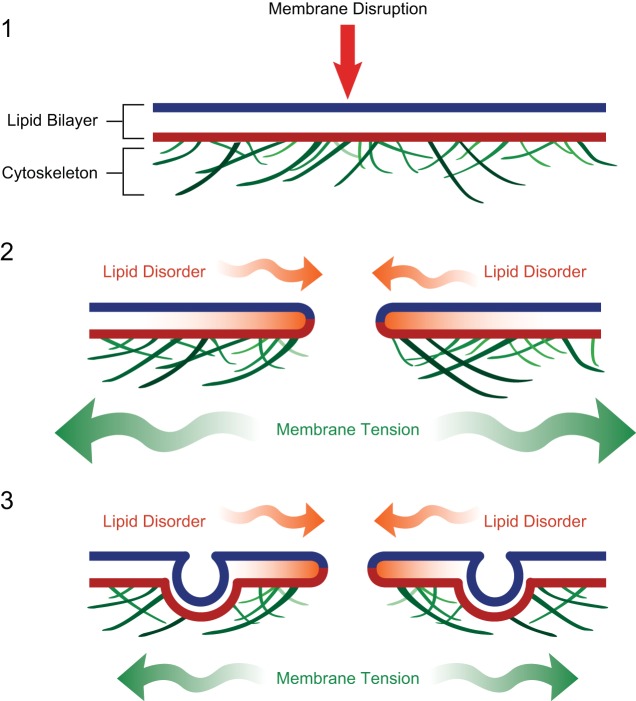
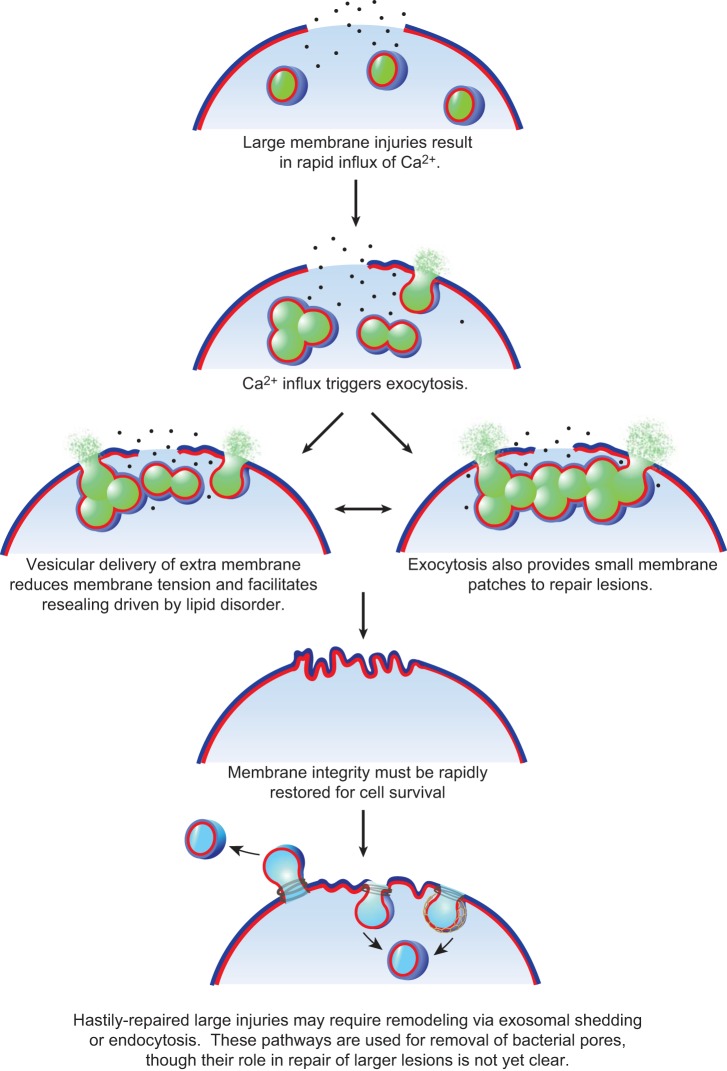
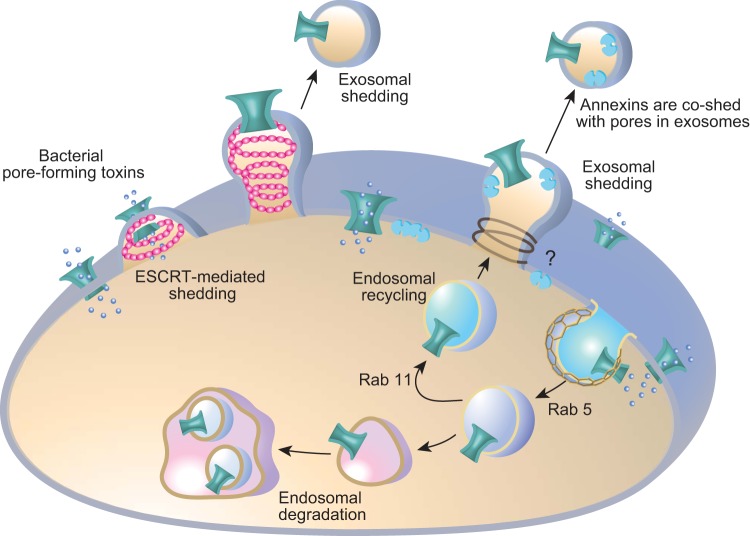
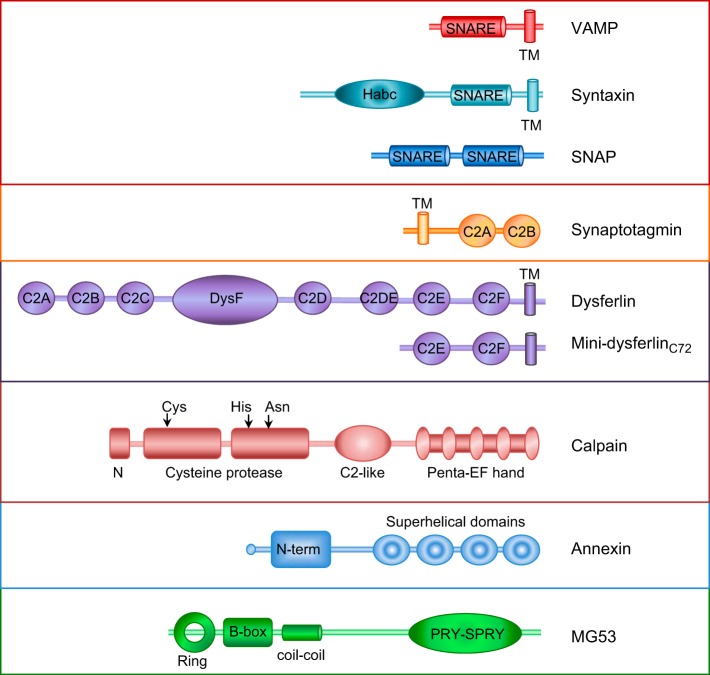
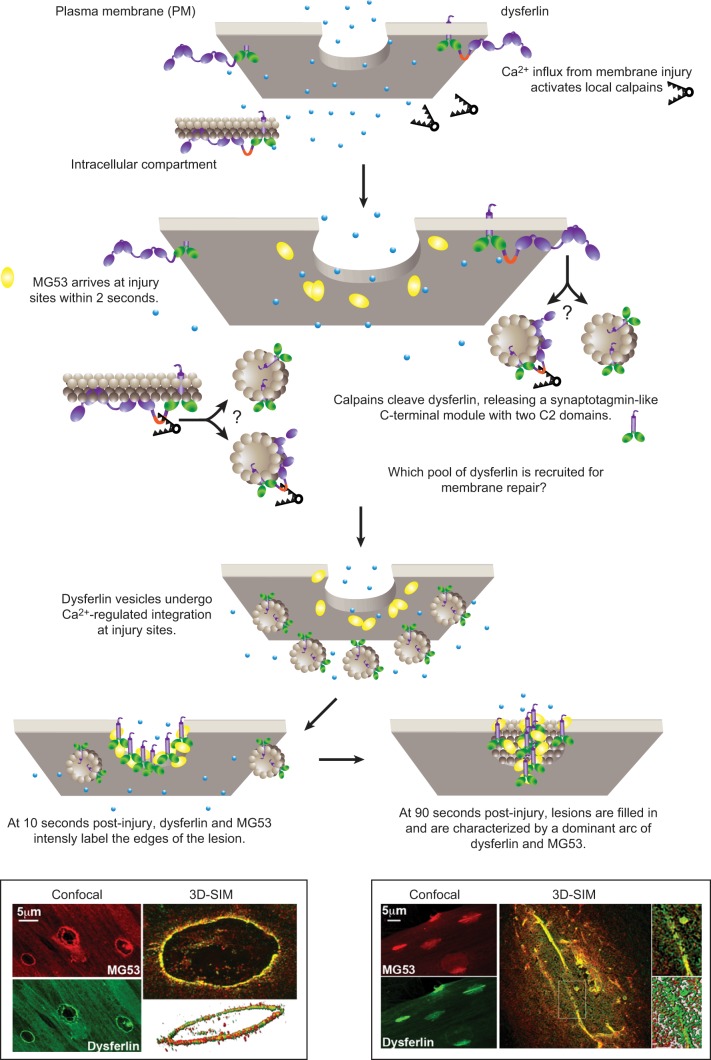
Eukaryotic cells have been confronted throughout their evolution with potentially lethal plasma membrane injuries, including those caused by osmotic stress, by infection from bacterial toxins and parasites, and by mechanical and ischemic stress. The wounded cell can survive if a rapid repair response is mounted that restores boundary integrity. Calcium has been identified as the key trigger to activate an effective membrane repair response that utilizes exocytosis and endocytosis to repair a membrane tear, or remove a membrane pore. We here review what is known about the cellular and molecular mechanisms of membrane repair, with particular emphasis on the relevance of repair as it relates to disease pathologies. Collective evidence reveals membrane repair employs primitive yet robust molecular machinery, such as vesicle fusion and contractile rings, processes evolutionarily honed for simplicity and success. Yet to be fully understood is whether core membrane repair machinery exists in all cells, or whether evolutionary adaptation has resulted in multiple compensatory repair pathways that specialize in different tissues and cells within our body.
Copyright © 2015 the American Physiological Society.
Figures
References
-
- Achanzar WE, Ward S. A nematode gene required for sperm vesicle fusion. J Cell Sci 110: 1073–1081, 1997. - PubMed
-
- Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxidants Redox Signal 12: 125–169, 2010. - PubMed
-
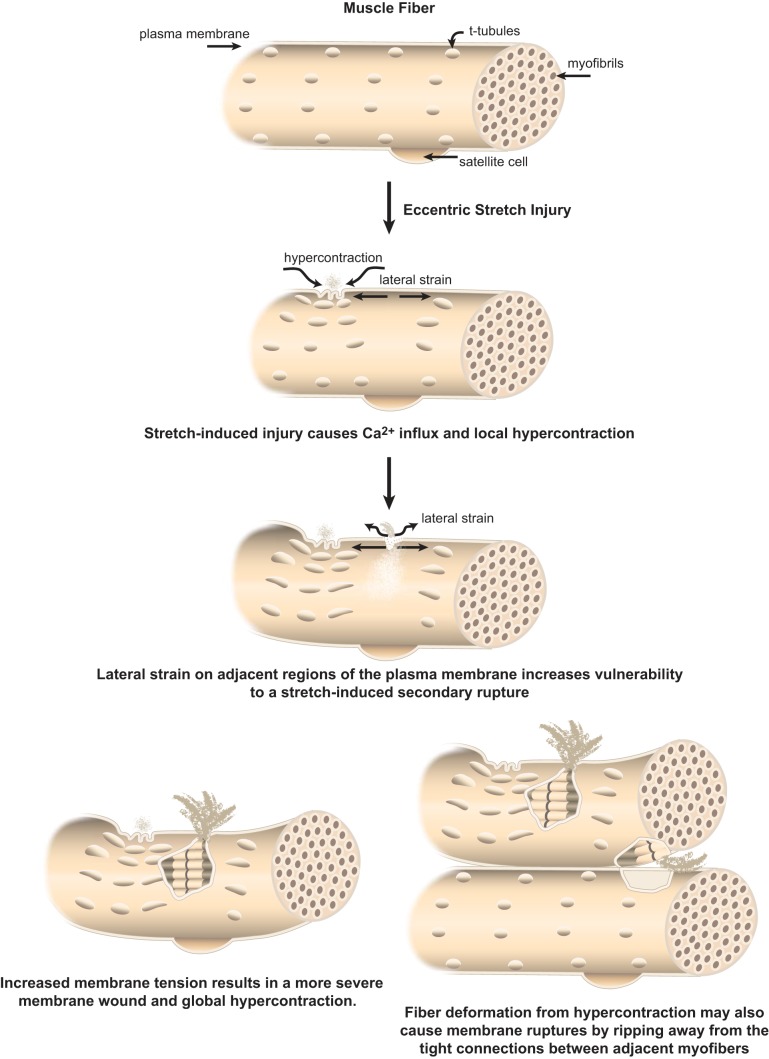
- Allen DG. Eccentric muscle damage: mechanisms of early reduction of force. Acta Physiol Scand 171: 311–319, 2001. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources