

MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities
- PMID: 26336640
- PMCID: PMC4556158
- DOI: 10.7717/peerj.1165
MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities
Abstract
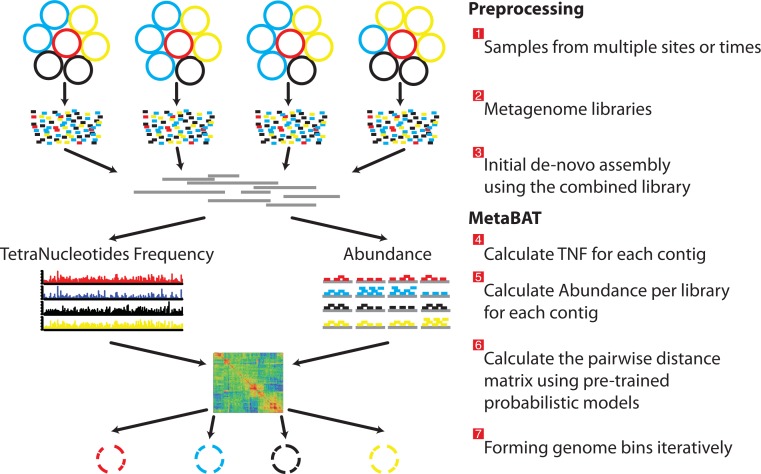
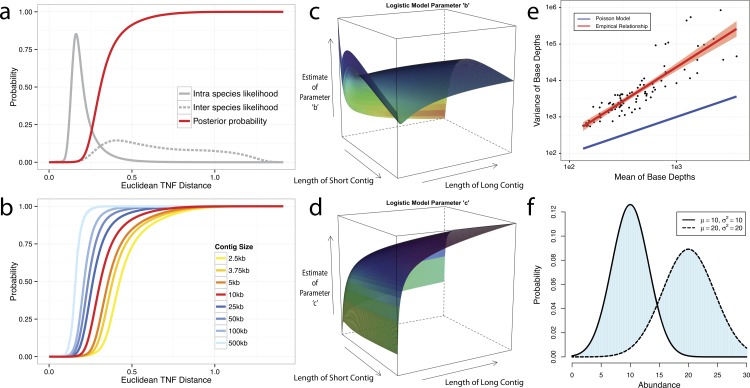
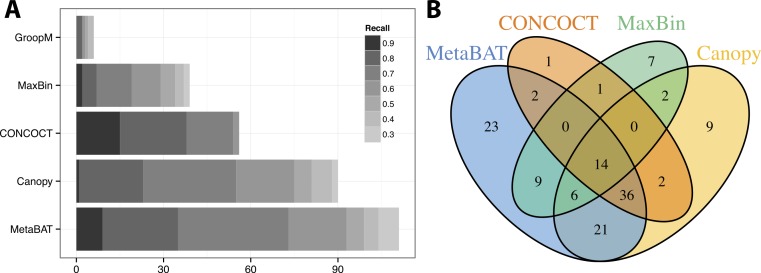
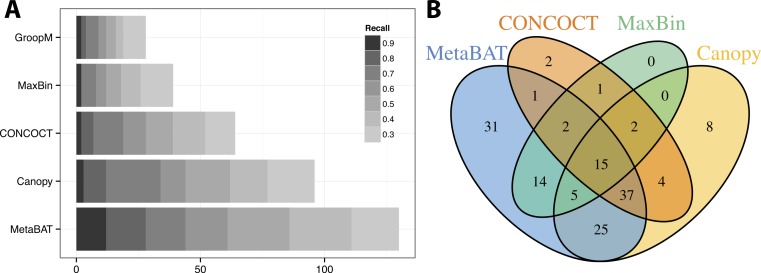
Grouping large genomic fragments assembled from shotgun metagenomic sequences to deconvolute complex microbial communities, or metagenome binning, enables the study of individual organisms and their interactions. Because of the complex nature of these communities, existing metagenome binning methods often miss a large number of microbial species. In addition, most of the tools are not scalable to large datasets. Here we introduce automated software called MetaBAT that integrates empirical probabilistic distances of genome abundance and tetranucleotide frequency for accurate metagenome binning. MetaBAT outperforms alternative methods in accuracy and computational efficiency on both synthetic and real metagenome datasets. It automatically forms hundreds of high quality genome bins on a very large assembly consisting millions of contigs in a matter of hours on a single node. MetaBAT is open source software and available at https://bitbucket.org/berkeleylab/metabat.
Keywords: MetaBAT; Metagenome binning.
Conflict of interest statement
The authors declare there are no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
