

Targeting protein aggregation for the treatment of degenerative diseases
- PMID: 26338154
- PMCID: PMC4628595
- DOI: 10.1038/nrd4593
Targeting protein aggregation for the treatment of degenerative diseases
Abstract
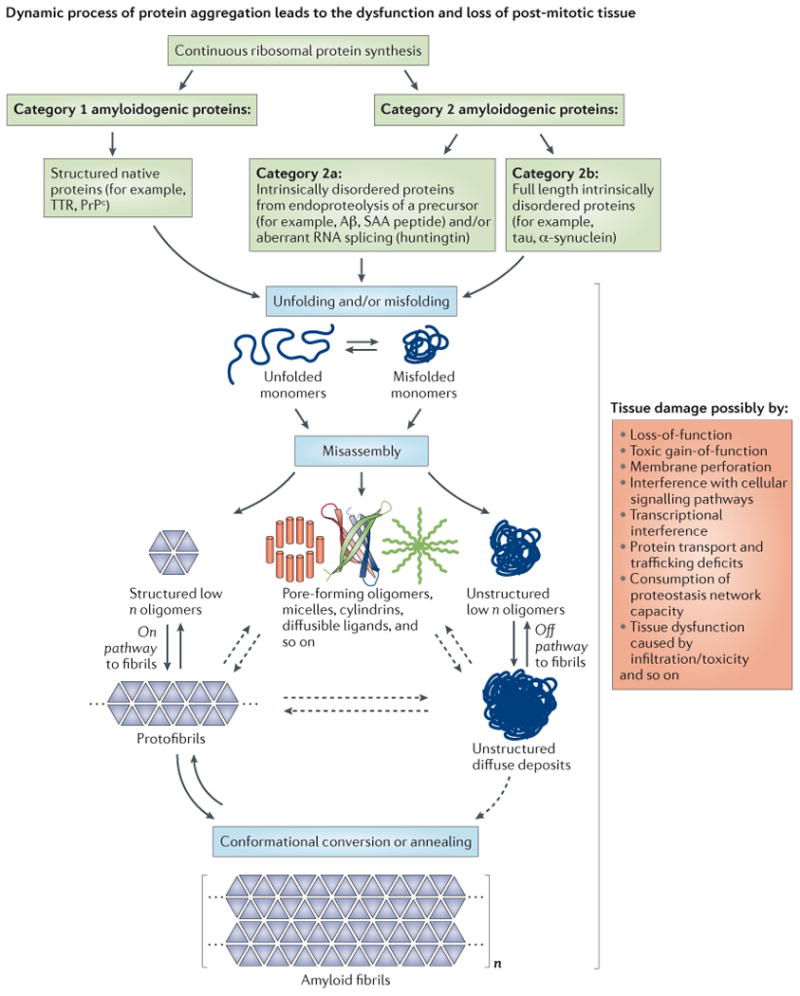
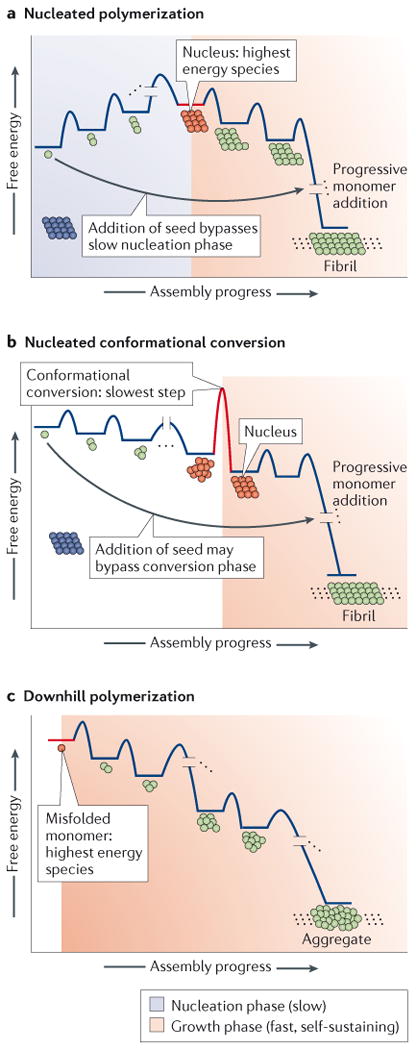
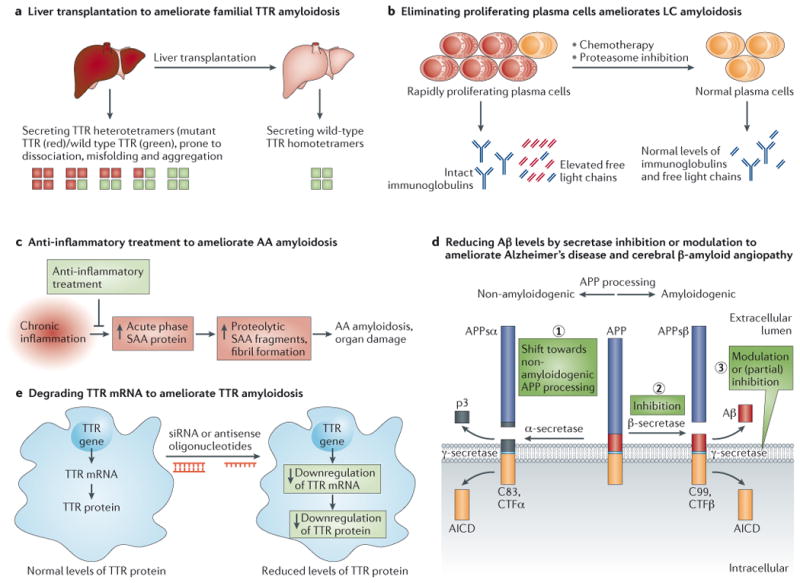
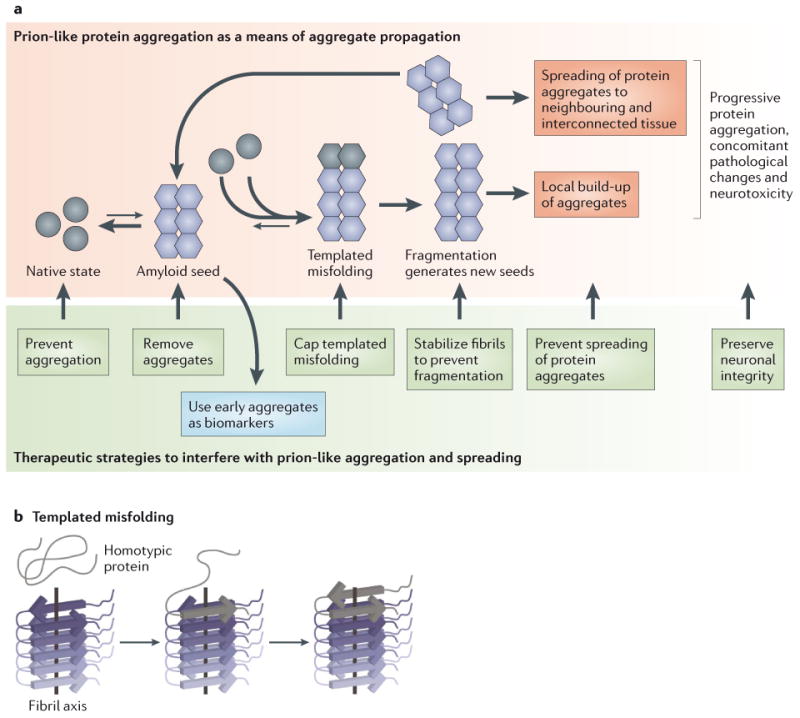
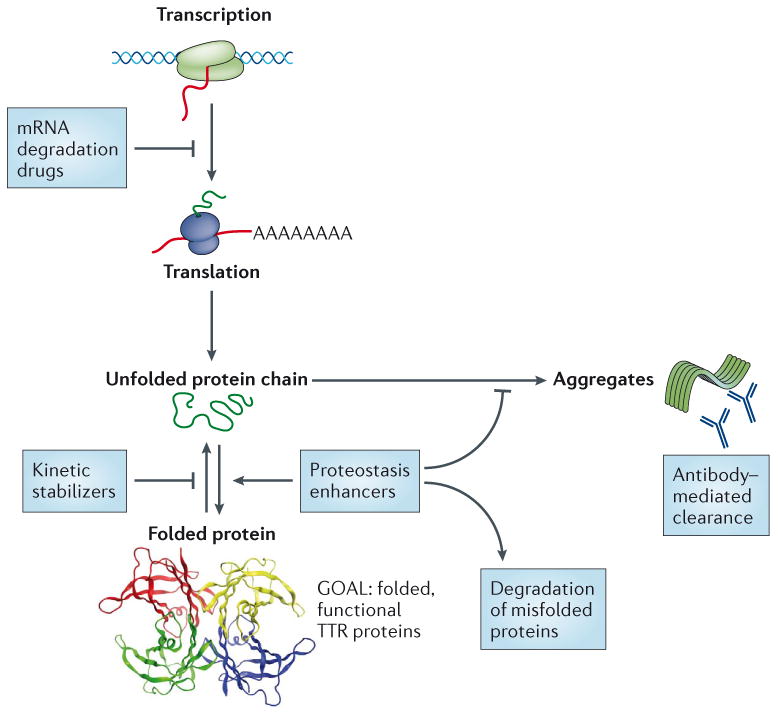
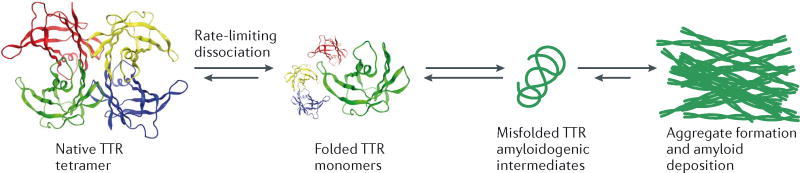
The aggregation of specific proteins is hypothesized to underlie several degenerative diseases, which are collectively known as amyloid disorders. However, the mechanistic connection between the process of protein aggregation and tissue degeneration is not yet fully understood. Here, we review current and emerging strategies to ameliorate aggregation-associated degenerative disorders, with a focus on disease-modifying strategies that prevent the formation of and/or eliminate protein aggregates. Persuasive pharmacological and genetic evidence now supports protein aggregation as the cause of postmitotic tissue dysfunction or loss. However, a more detailed understanding of the factors that trigger and sustain aggregate formation and of the structure-activity relationships underlying proteotoxicity is needed to develop future disease-modifying therapies.
Figures
References
-
- Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–27. - PubMed
-
- Glenner GG, Terry W, Harada M, Isersky C, Page D. Amyloid fibril proteins - proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150. &. - PubMed
-
- Linke RP, et al. Characteristics of a serum substance (SAA) antigenically related to amyloif fibril protein AA. Zeitschrift Fur Immunitats-Forschung Experimentelle Und Klinische Immunologie. 1975;150:219–219.
-
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–5. - PubMed
-
- Sipe JD, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21:221–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
