

A Disease Mutation Causing Episodic Ataxia Type I in the S1 Links Directly to the Voltage Sensor and the Selectivity Filter in Kv Channels
- PMID: 26338330
- PMCID: PMC6605306
- DOI: 10.1523/JNEUROSCI.1419-15.2015
A Disease Mutation Causing Episodic Ataxia Type I in the S1 Links Directly to the Voltage Sensor and the Selectivity Filter in Kv Channels
Abstract
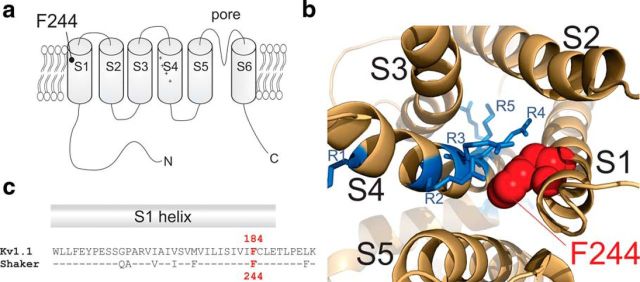
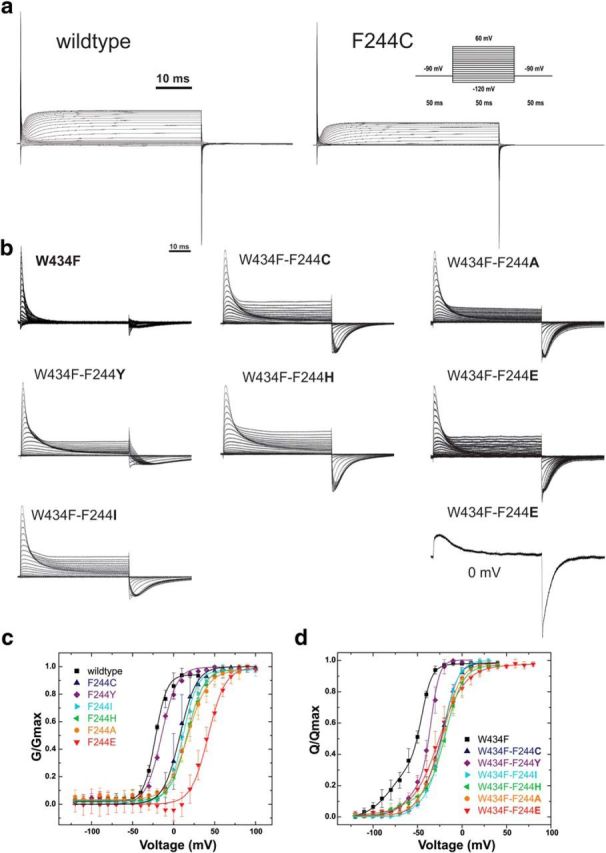
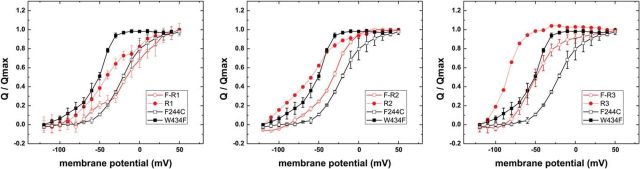
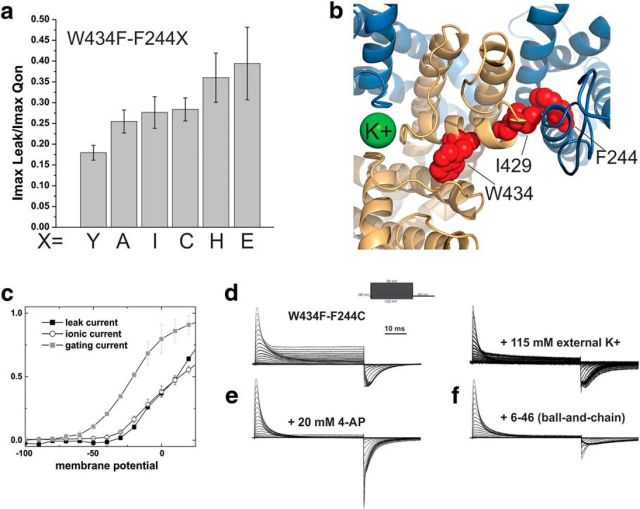
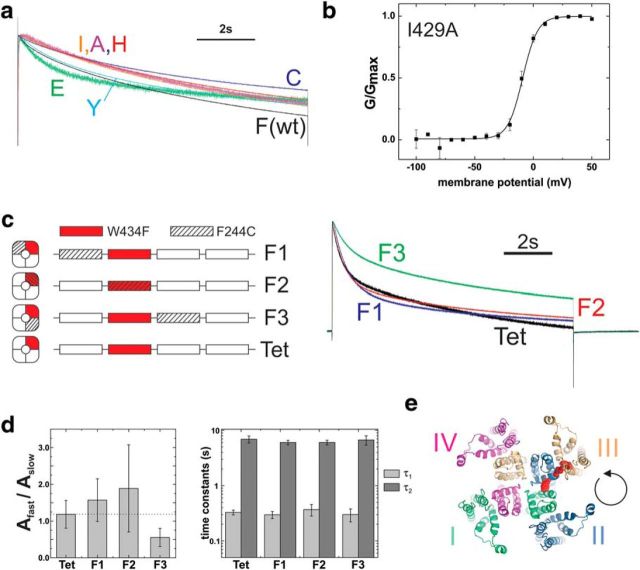
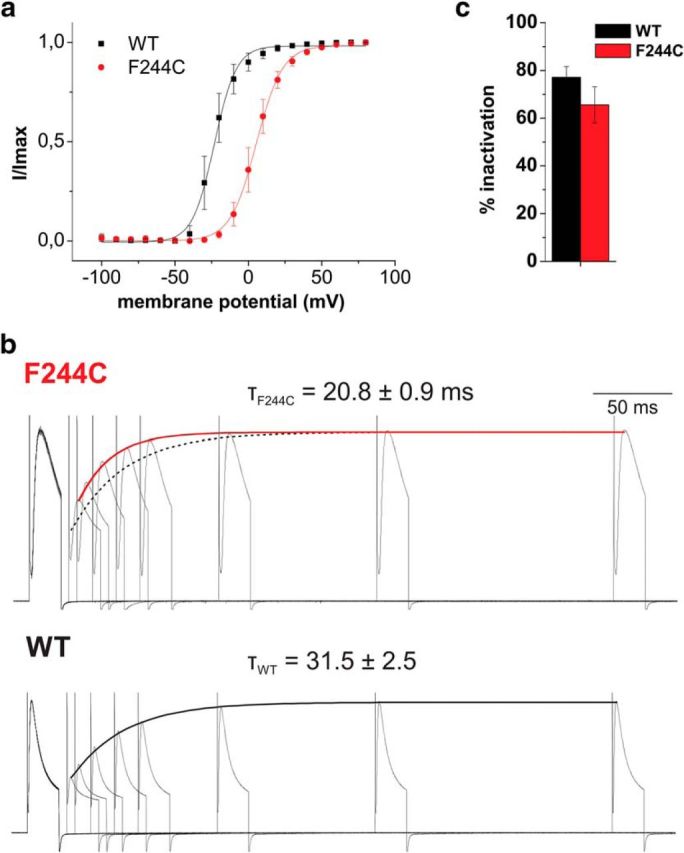
The mutation F184C in Kv1.1 leads to development of episodic ataxia type I (EA1). Although the mutation has been said to alter activation kinetics and to lower expression, we show here that the underlying molecular mechanisms may be more complex. Although F184 is positioned in the "peripheral" S1 helix, it occupies a central position in the 3D fold. We show in cut-open oocyte voltage-clamp recordings of gating and ionic currents of the Shaker Kv channel expressed in Xenopus oocytes that F184 not only interacts directly with the gating charges of the S4, but also creates a functional link to the selectivity filter of the neighboring subunit. This link leads to impaired fast and slow inactivation. The effect on fast inactivation is of an allosteric nature considering that fast inactivation is caused by a linked cytosolic ball peptide. The extensive effects of F184C provide a new mechanism underlying EA.
Significance statement: Episodic ataxia (EA) is an inherited disease that leads to occasional loss of motor control in combination with variable other symptoms such as vertigo or migraine. EA type I (EA1), studied here, is caused by mutations in a voltage-gated potassium channel that contributes to the generation of electrical signals in the brain. The mechanism by which mutations in voltage-gated potassium channels lead to EA is still unknown and there is no consistent pharmacological treatment. By studying in detail one disease-causing mutation in Kv1.1, we describe a novel molecular mechanism distinct from mechanisms described previously. This mechanism contributes to the understanding of potassium channel function in general and might lead to a better understanding of how EA develops.
Keywords: Kv channels; Xenopus oocytes; episodic ataxia; gating currents.
Copyright © 2015 the authors 0270-6474/15/3512198-09$15.00/0.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources