

Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data
- PMID: 26338770
- PMCID: PMC4673975
- DOI: 10.1093/bioinformatics/btv488
Benchmark analysis of algorithms for determining and quantifying full-length mRNA splice forms from RNA-seq data
Abstract
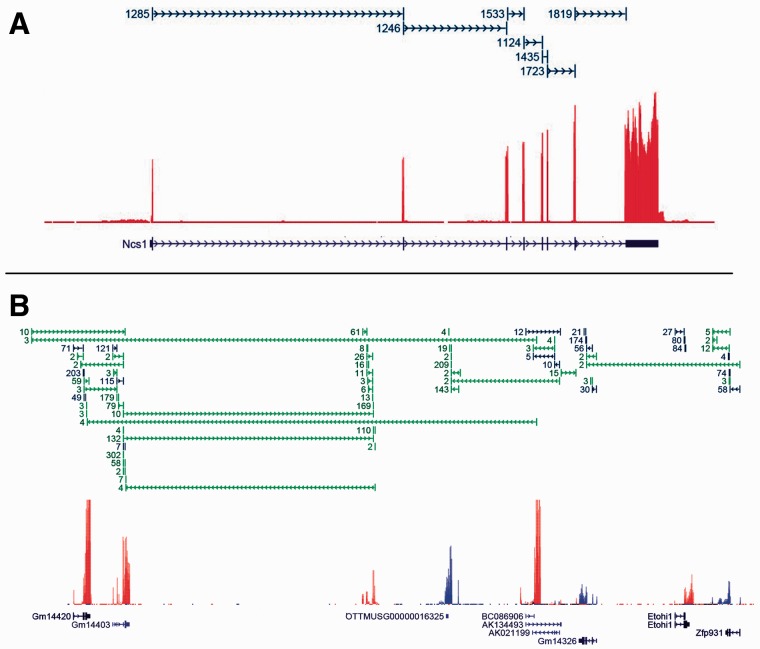
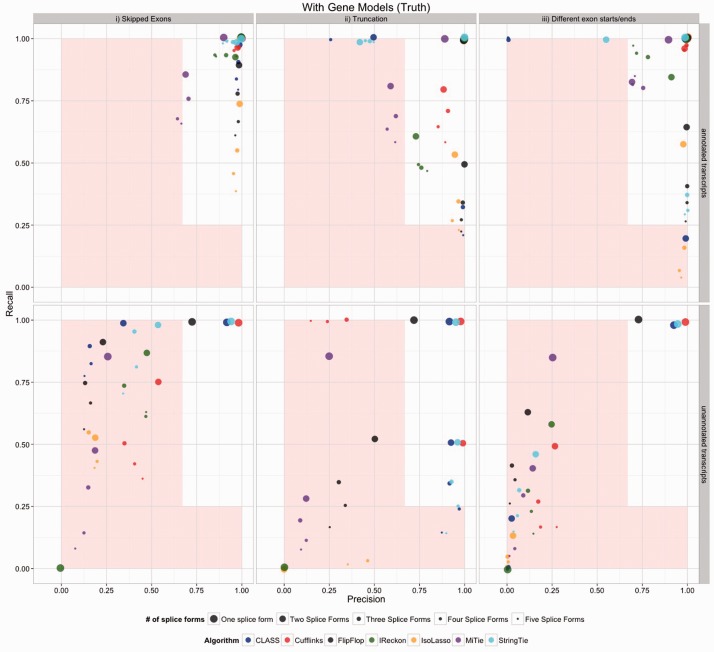
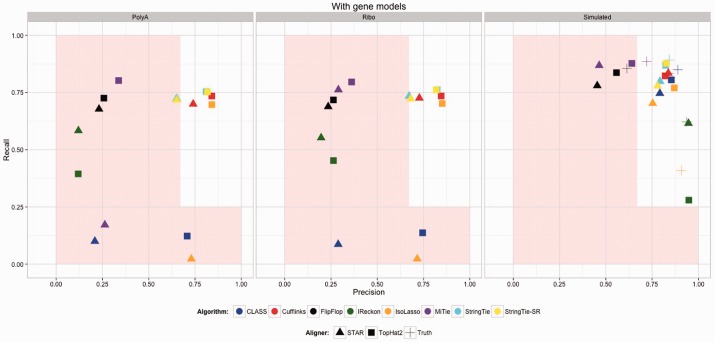
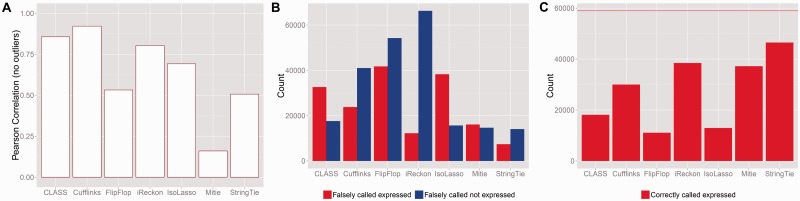
Motivation: Because of the advantages of RNA sequencing (RNA-Seq) over microarrays, it is gaining widespread popularity for highly parallel gene expression analysis. For example, RNA-Seq is expected to be able to provide accurate identification and quantification of full-length splice forms. A number of informatics packages have been developed for this purpose, but short reads make it a difficult problem in principle. Sequencing error and polymorphisms add further complications. It has become necessary to perform studies to determine which algorithms perform best and which if any algorithms perform adequately. However, there is a dearth of independent and unbiased benchmarking studies. Here we take an approach using both simulated and experimental benchmark data to evaluate their accuracy.
Results: We conclude that most methods are inaccurate even using idealized data, and that no method is highly accurate once multiple splice forms, polymorphisms, intron signal, sequencing errors, alignment errors, annotation errors and other complicating factors are present. These results point to the pressing need for further algorithm development.
Availability and implementation: Simulated datasets and other supporting information can be found at http://bioinf.itmat.upenn.edu/BEERS/bp2.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2015. Published by Oxford University Press.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
