

A predictive modeling approach for cell line-specific long-range regulatory interactions
- PMID: 26338778
- PMCID: PMC4605315
- DOI: 10.1093/nar/gkv865
A predictive modeling approach for cell line-specific long-range regulatory interactions
Erratum in
-
A predictive modeling approach for cell line-specific long-range regulatory interactions.Nucleic Acids Res. 2016 Feb 29;44(4):1977-8. doi: 10.1093/nar/gkv1181. Epub 2015 Nov 5. Nucleic Acids Res. 2016. PMID: 26546512 Free PMC article. No abstract available.
Abstract
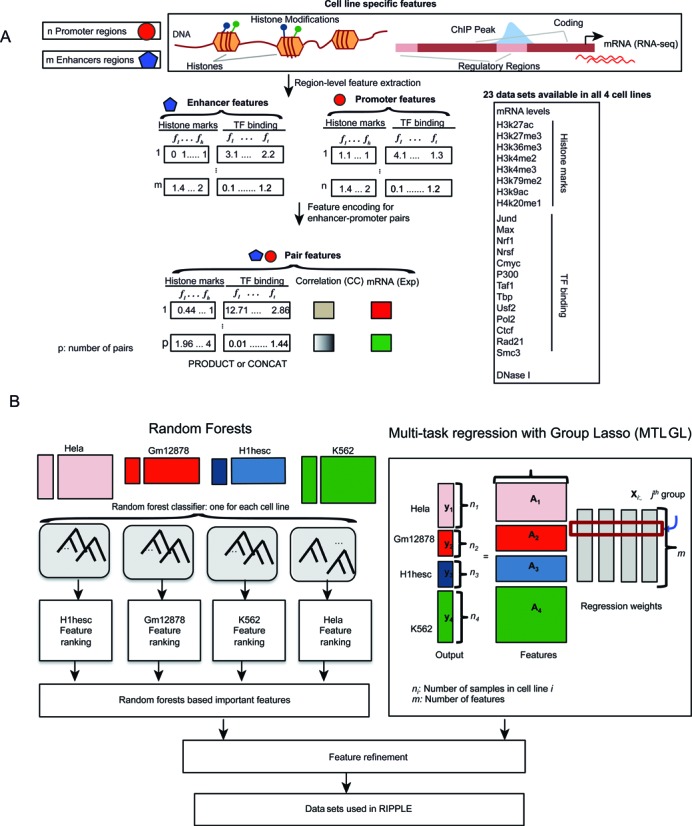
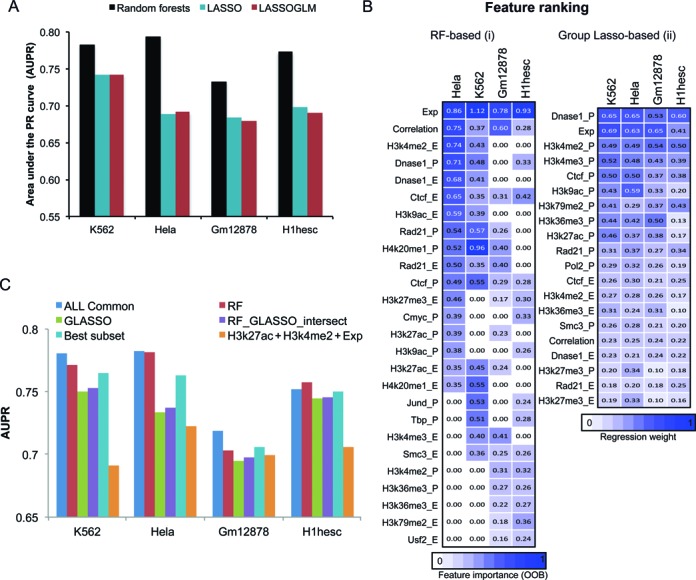
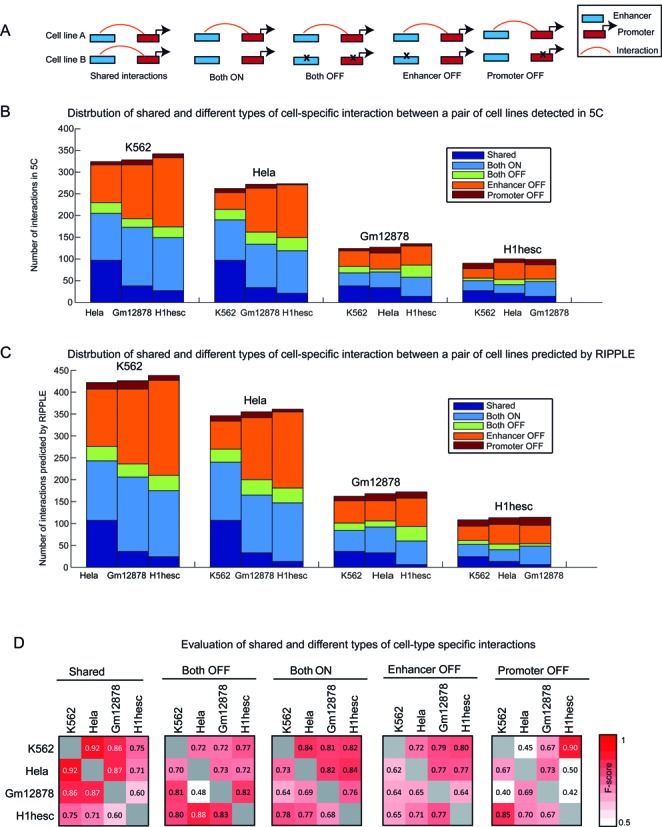
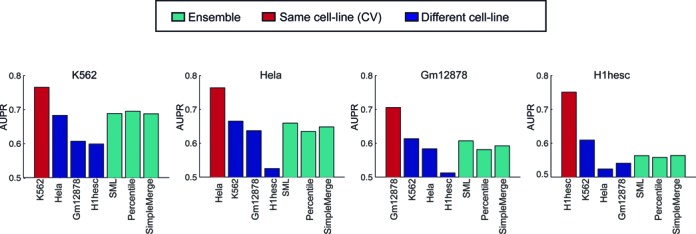
Long range regulatory interactions among distal enhancers and target genes are important for tissue-specific gene expression. Genome-scale identification of these interactions in a cell line-specific manner, especially using the fewest possible datasets, is a significant challenge. We develop a novel computational approach, Regulatory Interaction Prediction for Promoters and Long-range Enhancers (RIPPLE), that integrates published Chromosome Conformation Capture (3C) data sets with a minimal set of regulatory genomic data sets to predict enhancer-promoter interactions in a cell line-specific manner. Our results suggest that CTCF, RAD21, a general transcription factor (TBP) and activating chromatin marks are important determinants of enhancer-promoter interactions. To predict interactions in a new cell line and to generate genome-wide interaction maps, we develop an ensemble version of RIPPLE and apply it to generate interactions in five human cell lines. Computational validation of these predictions using existing ChIA-PET and Hi-C data sets showed that RIPPLE accurately predicts interactions among enhancers and promoters. Enhancer-promoter interactions tend to be organized into subnetworks representing coordinately regulated sets of genes that are enriched for specific biological processes and cis-regulatory elements. Overall, our work provides a systematic approach to predict and interpret enhancer-promoter interactions in a genome-wide cell-type specific manner using a few experimentally tractable measurements.
© The Author(s) 2015. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
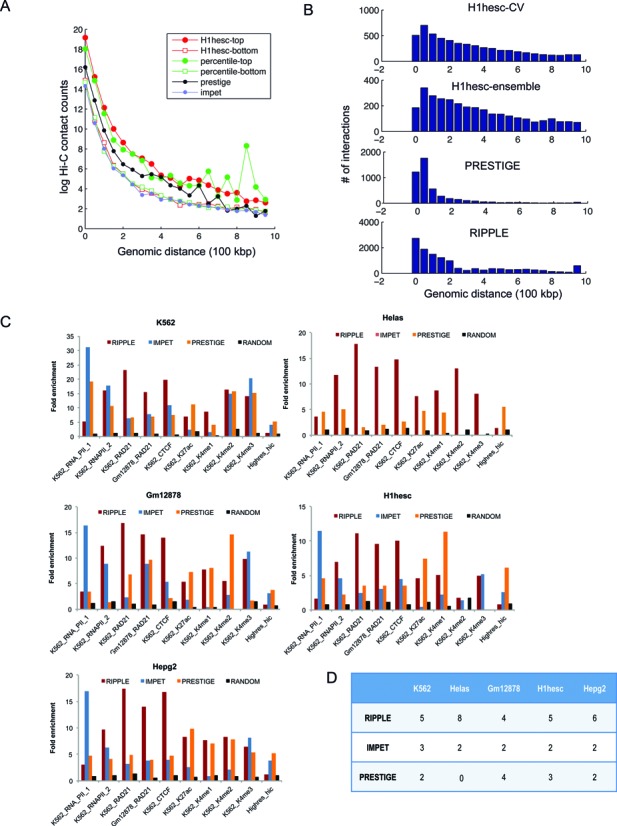

References
-
- Krijger P.H., de Laat W. Identical cells with different 3D genomes; cause and consequences. Curr. Opin. Genet. Dev. 2013;23:191–196. - PubMed
-
- de Laat W., Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. - PubMed
-
- Dekker J., Rippe K., Dekker M., Kleckner N. Capturing Chromosome Conformation. Science. 2002;295:1306–1311. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
