

Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry
- PMID: 26342526
- PMCID: PMC4699615
- DOI: 10.1016/j.jaci.2015.06.048
Genetic predisposition to hemophagocytic lymphohistiocytosis: Report on 500 patients from the Italian registry
Abstract
Background: Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening disease affecting mostly children but also adults and characterized by hyperinflammatory features. A subset of patients, referred to as having familial hemophagocytic lymphohistiocytosis (FHL), have various underlying genetic abnormalities, the frequencies of which have not been systematically determined previously.
Objective: This work aims to further our understanding of the pathogenic bases of this rare condition based on an analysis of our 25 years of experience.
Methods: From our registry, we have analyzed a total of 500 unselected patients with HLH.
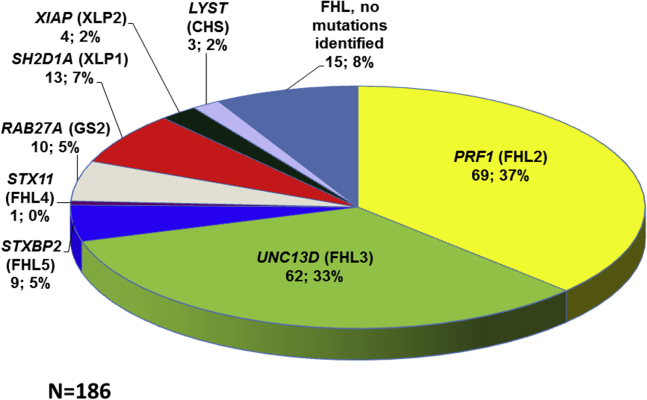
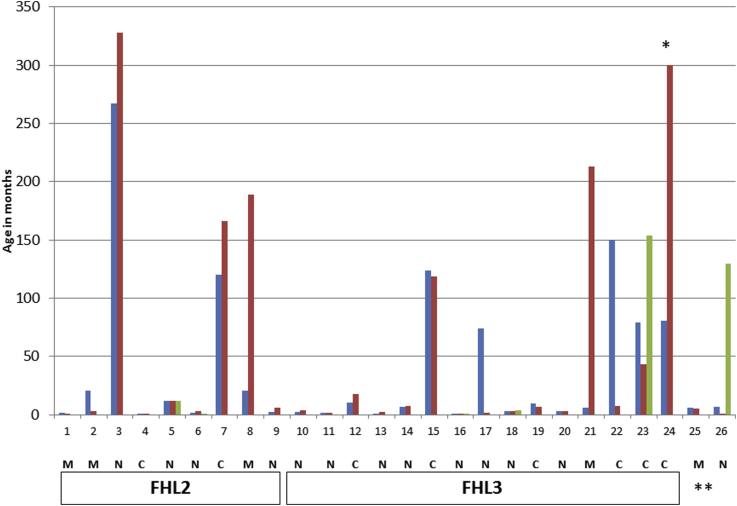
Results: Biallelic pathogenic mutations defining FHL were found in 171 (34%) patients; the proportion of FHL was much higher (64%) in patients given a diagnosis during the first year of life. Taken together, mutations of the genes PRF1 (FHL2) and UNC13D (FHL3) accounted for 70% of cases of FHL. Overall, a genetic diagnosis was possible in more than 90% of our patients with FHL. Perforin expression and the extent of degranulation have been more useful for diagnosing FHL than hemophagocytosis and the cytotoxicity assay. Of 281 (56%) patients classified as having "sporadic" HLH, 43 had monoallelic mutations in one of the FHL-defining genes. Given this gene dosage effect, FHL is not strictly recessive.
Conclusion: We suggest that the clinical syndrome HLH generally results from the combined effects of an exogenous trigger and genetic predisposition. Within this combination, different weights of exogenous and genetic factors account for the wide disease spectrum that ranges from HLH secondary to severe infection to FHL.
Keywords: Hemophagocytic lymphohistiocytosis; PRF1; UNC13D; immunologic tests.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Bodley-Scott R., Robb-Smith A.H.T. Histiocytic medullary reticulosis. Lancet. 1939;234(6047):194–198.
-
- Arico M., Janka G., Fischer A., Henter J.I., Blanche S., Elinder G. Haemophagocytic lymphohistiocytosis: report of 122 children from the International Registry. Leukemia. 1996;10:197–203. - PubMed
-
- Janka G., zur Stadt U. Familial and acquired hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2005:82–88. - PubMed
-
- Stepp S.E., Dufourcq-Lagelouse R., Le Deist F., Bhawan S., Certain S., Mathew P.A. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–1959. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
