

BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition
- PMID: 26343582
- PMCID: PMC4894664
- DOI: 10.1016/j.ccell.2015.08.001
BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition
Abstract
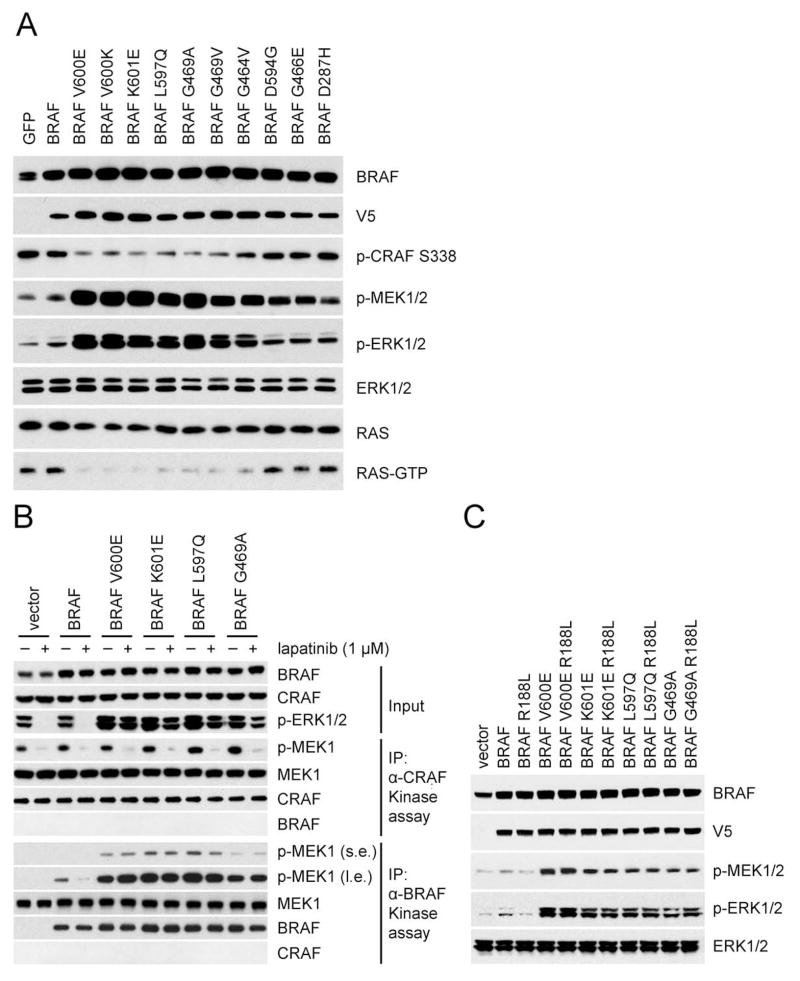
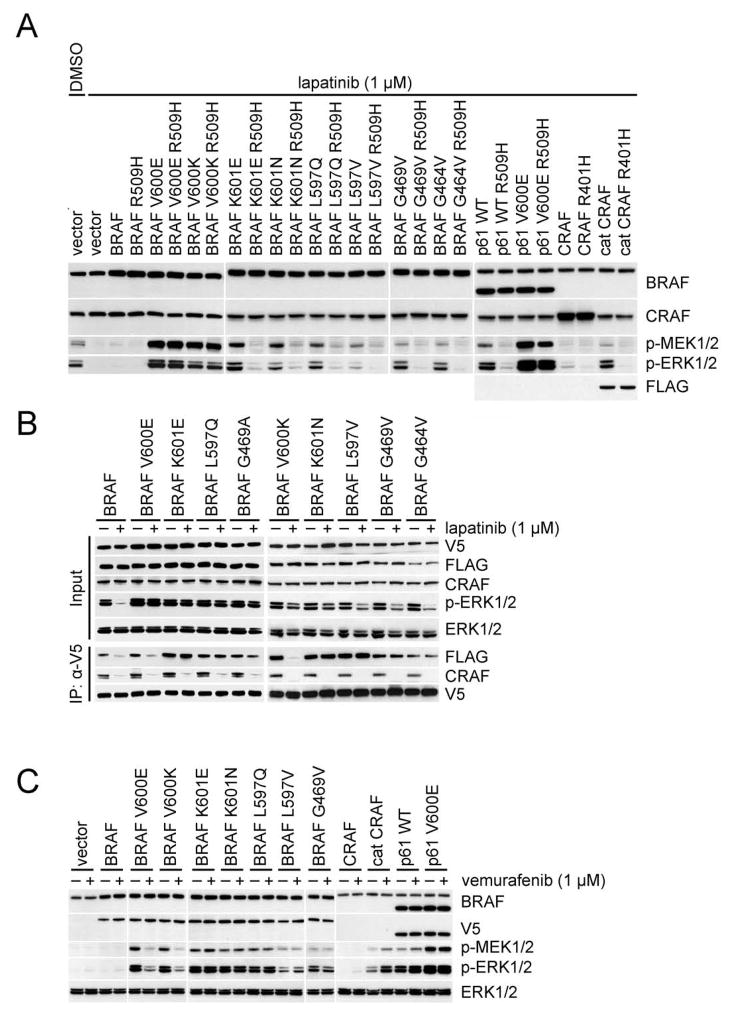
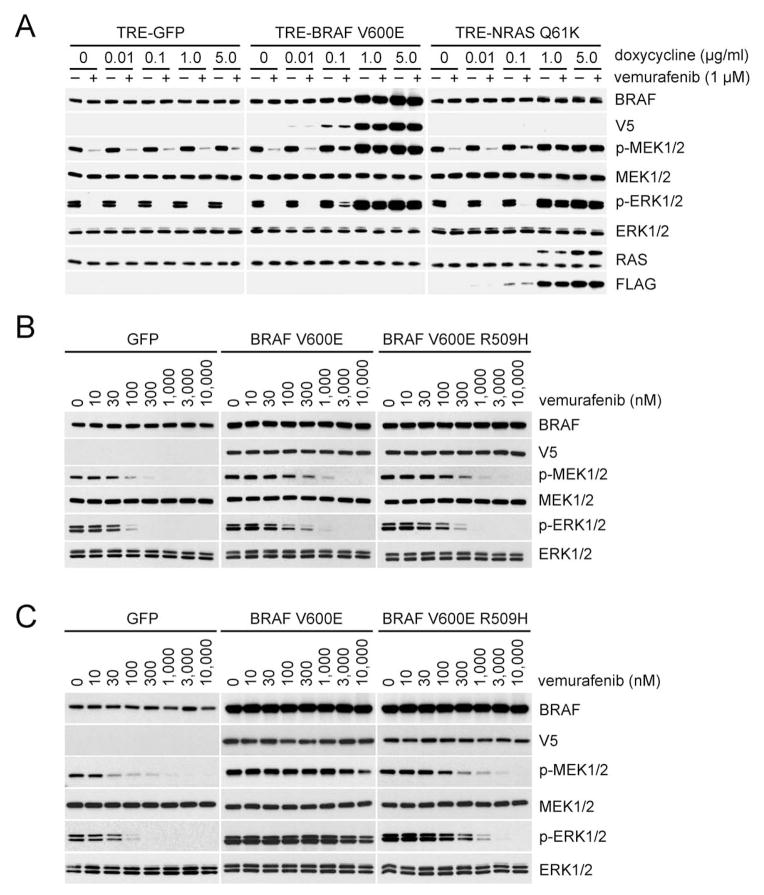
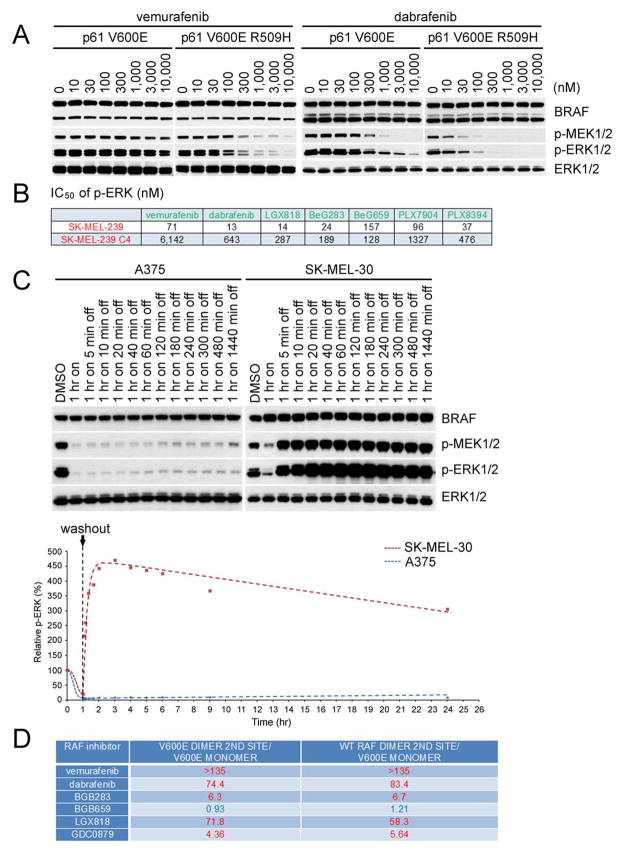
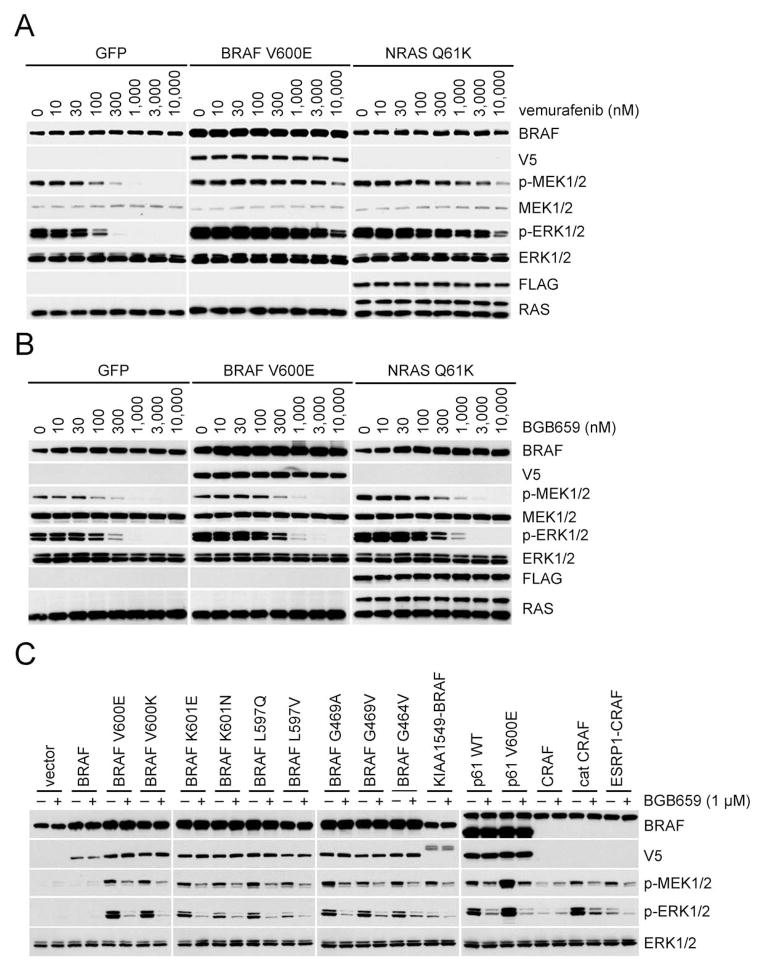
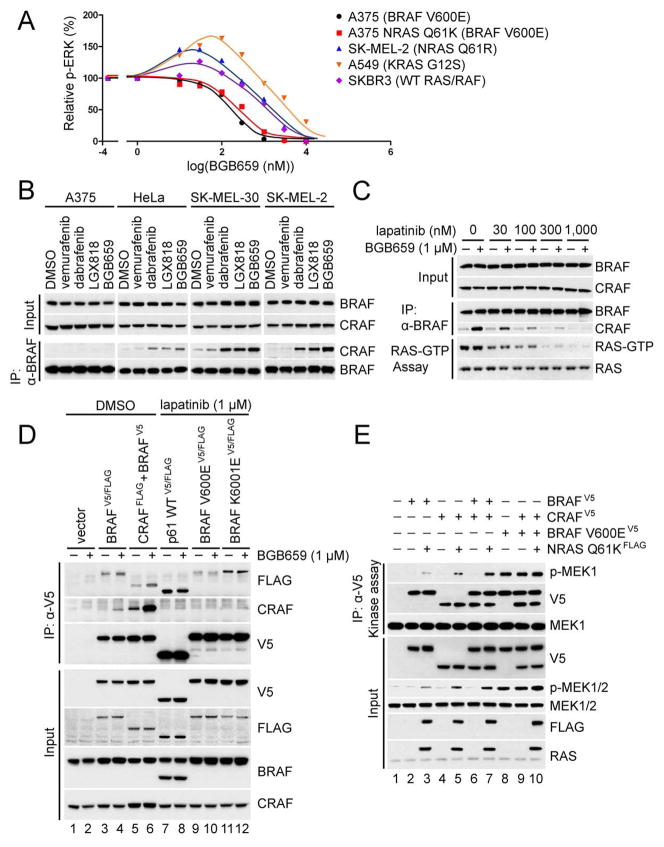
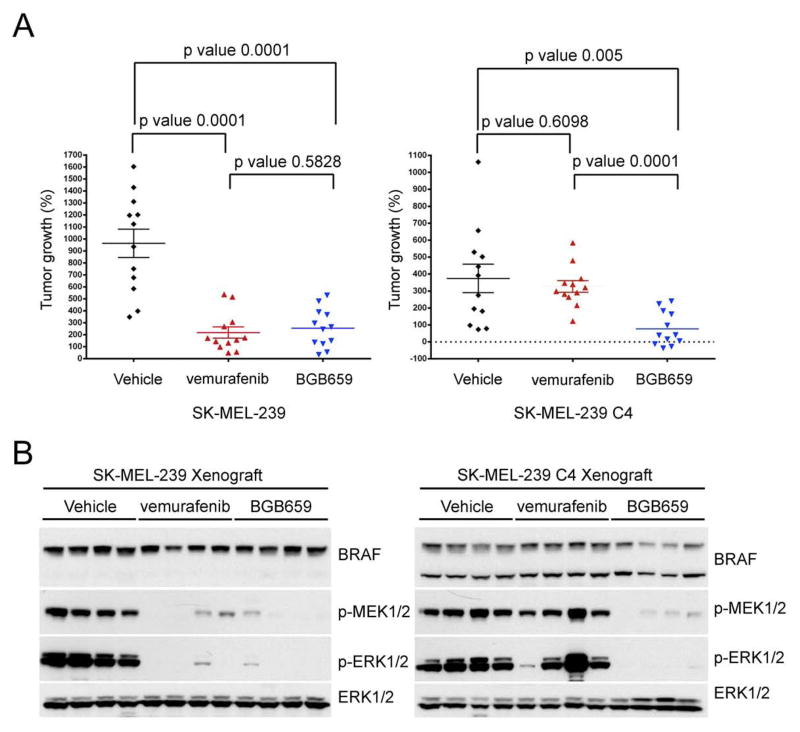
ERK signaling requires RAS-induced RAF dimerization and is limited by feedback. Activated BRAF mutants evade feedback inhibition of RAS by either of two mechanisms. BRAF V600 mutants are activated monomers when RAS activity is low; all other activating BRAF mutants function as constitutive RAS-independent dimers. RAF inhibitors effectively inhibit mutant monomers, but not dimers; their binding to one site in the dimer significantly reduces their affinity for the second. Tumors with non-V600E BRAF mutants are insensitive to these drugs, and increased expression of BRAF V600E dimers causes acquired resistance. A compound that equally inhibits both sites of mutant RAF dimers inhibits tumors driven by either class of mutants or those BRAF V600E tumors with dimer-dependent acquired resistance to monomer-specific inhibitors.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Path Forward for RAF Therapies: Inhibition of Monomers and Dimers.Cancer Cell. 2015 Sep 14;28(3):279-81. doi: 10.1016/j.ccell.2015.08.006. Cancer Cell. 2015. PMID: 26373275 Free PMC article.
References
-
- Avraham R, Yarden Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol. 2011;12:104–117. - PubMed
-
- Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27:689–696. - PubMed
-
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. - PMC - PubMed
-
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
