

ConStrains identifies microbial strains in metagenomic datasets
- PMID: 26344404
- PMCID: PMC4676274
- DOI: 10.1038/nbt.3319
ConStrains identifies microbial strains in metagenomic datasets
Abstract
An important fraction of microbial diversity is harbored in strain individuality, so identification of conspecific bacterial strains is imperative for improved understanding of microbial community functions. Limitations in bioinformatics and sequencing technologies have to date precluded strain identification owing to difficulties in phasing short reads to faithfully recover the original strain-level genotypes, which have highly similar sequences. We present ConStrains, an open-source algorithm that identifies conspecific strains from metagenomic sequence data and reconstructs the phylogeny of these strains in microbial communities. The algorithm uses single-nucleotide polymorphism (SNP) patterns in a set of universal genes to infer within-species structures that represent strains. Applying ConStrains to simulated and host-derived datasets provides insights into microbial community dynamics.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
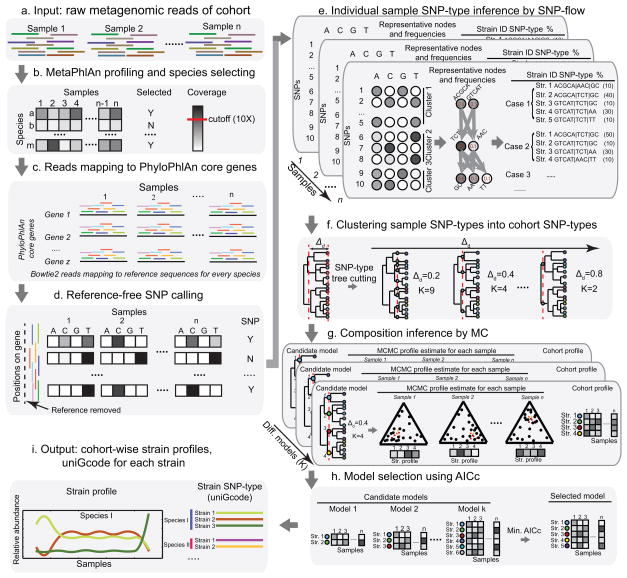
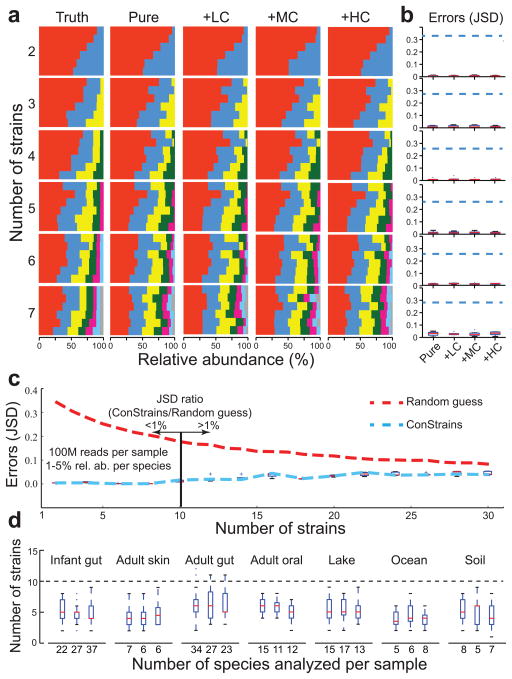
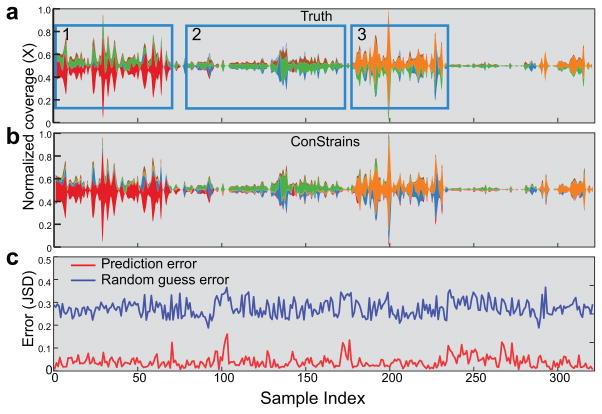
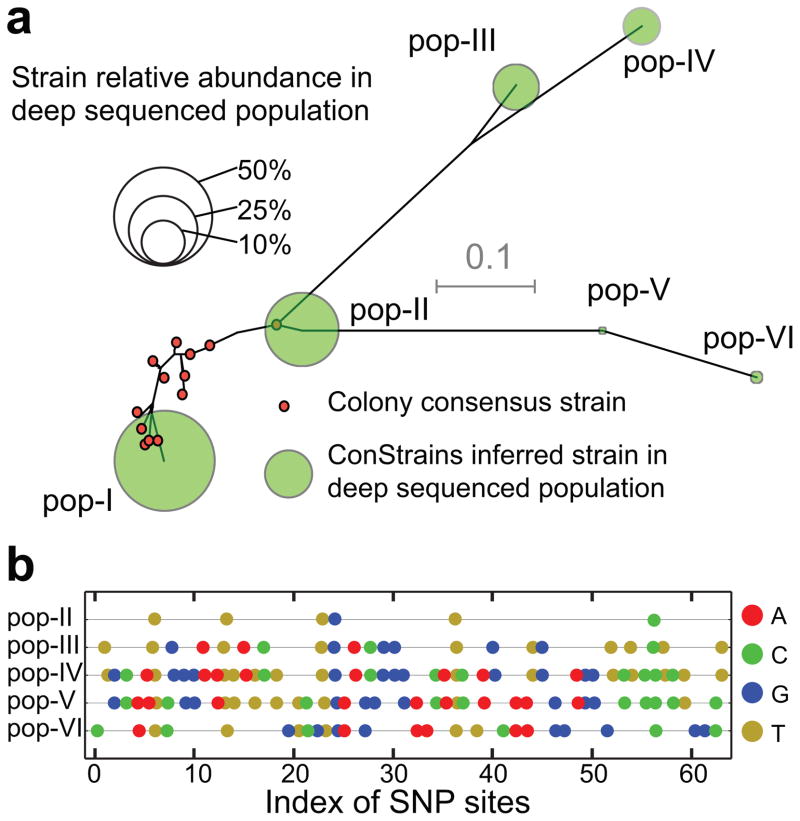
References
-
- Sunagawa S, et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat Methods. 2013;10:1196–1199. - PubMed
-
- Nielsen HB, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–828. - PubMed
