

Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease
- PMID: 26345314
- PMCID: PMC4554398
- DOI: 10.2147/DDDT.S77760
Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease
Erratum in
-
Erratum: Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease [Corrigendum].Drug Des Devel Ther. 2015 Sep 11;9:5213. doi: 10.2147/DDDT.S95612. eCollection 2015. Drug Des Devel Ther. 2015. PMID: 26388688 Free PMC article.
Abstract
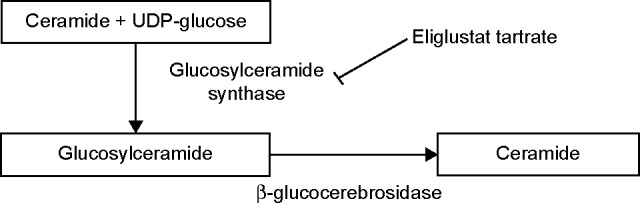
The purpose of this article is to review eliglustat tartrate, a substrate reduction therapy, for the treatment of Gaucher disease type 1 (GD1). GD is an rare inborn error of metabolism caused by accumulation of lipid substrates such as glucosylceramide within the monocyte-macrophage system that affects the body by causing enlargement of the spleen and liver, destruction of bone, and abnormalities of the lungs and blood, such as anemia, thrombocytopenia, and leukopenia. GD is classified into three types: GD1, a chronic and non-neuronopathic disease accounting for 95% of GD cases; and types 2 and 3 (GD2 GD3) which are more progressive diseases with no approved drugs available at this time. Treatment options for GD1 include enzyme replacement therapy and substrate reduction therapy. Eliglustat works by inhibiting UDP-glucosylceramide synthase, the first enzyme that catalyzes the biosynthesis of glycosphingolipids, thus reducing the load of glucosylceramide influx into the lysosome. Eliglustat was approved by the US Food and Drug Administration after three Phase I, two Phase II, and two Phase III clinical trials. The dose of eliglustat is 84 mg twice a day or once daily depending on the cytochrome P450 2D6 genotype of the patient.
Keywords: Gaucher disease; eliglustat tartrate; glucocerebrosidase; glucosylceramide synthase; substrate reduction therapy.
Figures
Similar articles
-
Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe.Eur J Intern Med. 2017 Jan;37:25-32. doi: 10.1016/j.ejim.2016.07.011. Epub 2016 Aug 10. Eur J Intern Med. 2017. PMID: 27522145 Review.
-
A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1.Blood. 2010 Aug 12;116(6):893-9. doi: 10.1182/blood-2010-03-273151. Epub 2010 May 3. Blood. 2010. PMID: 20439622 Free PMC article. Clinical Trial.
-
Eliglustat tartrate, an orally active glucocerebroside synthase inhibitor for the potential treatment of Gaucher disease and other lysosomal storage diseases.Curr Opin Investig Drugs. 2010 Oct;11(10):1169-81. Curr Opin Investig Drugs. 2010. PMID: 20872320 Review.
-
Gaucher disease and its treatment options.Ann Pharmacother. 2013 Sep;47(9):1182-93. doi: 10.1177/1060028013500469. Ann Pharmacother. 2013. PMID: 24259734 Review.
-
Budget Impact Analysis of Eliglustat for the Treatment of Gaucher Disease Type 1 in the United States.J Manag Care Spec Pharm. 2018 Oct;24(10):1002-1008. doi: 10.18553/jmcp.2018.24.10.1002. J Manag Care Spec Pharm. 2018. PMID: 30247105 Free PMC article.
Cited by
-
The Shiga Toxin Receptor Globotriaosylceramide as Therapeutic Target in Shiga Toxin E. coli Mediated HUS.Microorganisms. 2021 Oct 16;9(10):2157. doi: 10.3390/microorganisms9102157. Microorganisms. 2021. PMID: 34683478 Free PMC article.
-
Adeno-associated virus expressing a blood-brain barrier-penetrating enzyme improves GM1 gangliosidosis in a preclinical model.J Clin Invest. 2025 Apr 8;135(12):e180724. doi: 10.1172/JCI180724. eCollection 2025 Jun 16. J Clin Invest. 2025. PMID: 40198143 Free PMC article.
-
Pharmacotherapy of Gaucher Disease: Current and Future Options.P T. 2018 May;43(5):274-309. P T. 2018. PMID: 29719368 Free PMC article.
-
Current and emerging pharmacotherapy for Gaucher disease in pediatric populations.Expert Opin Pharmacother. 2021 Aug;22(11):1489-1503. doi: 10.1080/14656566.2021.1902989. Epub 2021 Mar 25. Expert Opin Pharmacother. 2021. PMID: 33711910 Free PMC article.
-
Comprehensive genome based analysis of Vibrio parahaemolyticus for identifying novel drug and vaccine molecules: Subtractive proteomics and vaccinomics approach.PLoS One. 2020 Aug 19;15(8):e0237181. doi: 10.1371/journal.pone.0237181. eCollection 2020. PLoS One. 2020. PMID: 32813697 Free PMC article.
References
-
- Mehta A. Epidemiology and natural history of Gaucher’s disease. Eur J Intern Med. 2006;17:S2–S5. - PubMed
-
- Goker-Alpan O. Therapeutic approaches to bone pathology in Gaucher disease: past, present and future. Mol Genet Metab. 2011;104:438–447. - PubMed
-
- Conradi NG, Kalimo H, Sourander P. Reactions of vessel walls and brain parenchyma to the accumulation of Gaucher cells in the Norrbottnian type (type III) of Gaucher disease. Acta Neuropathol. 1988;75:385–390. - PubMed
-
- Grabowski GA. Gaucher disease: lessons from a decade of therapy. J Pediatr. 2004;144:S15–S19. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
