

Role of the nucleus in apoptosis: signaling and execution
- PMID: 26346492
- PMCID: PMC11113907
- DOI: 10.1007/s00018-015-2031-y
Role of the nucleus in apoptosis: signaling and execution
Abstract
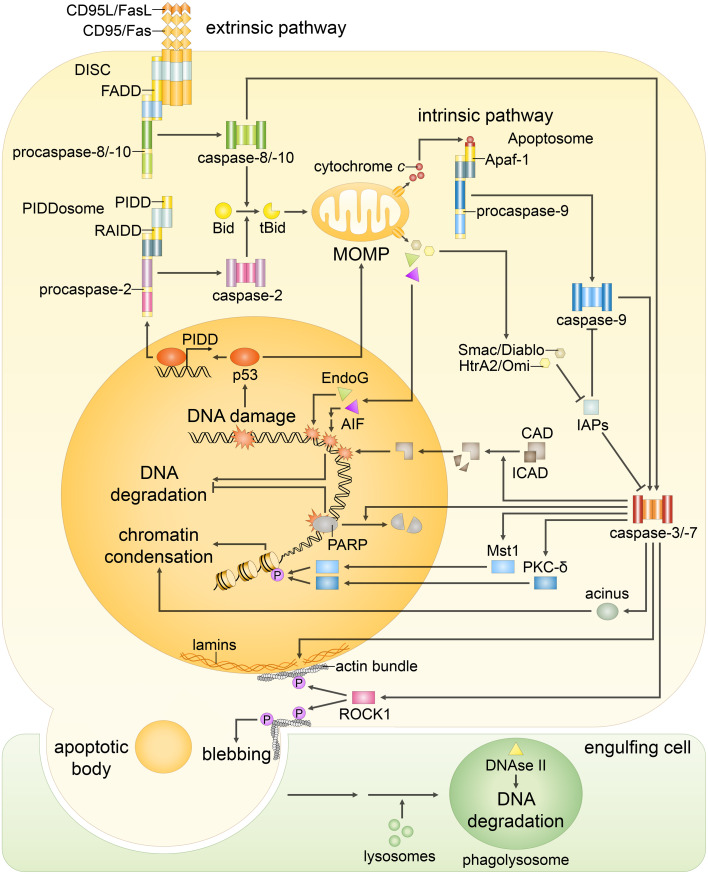
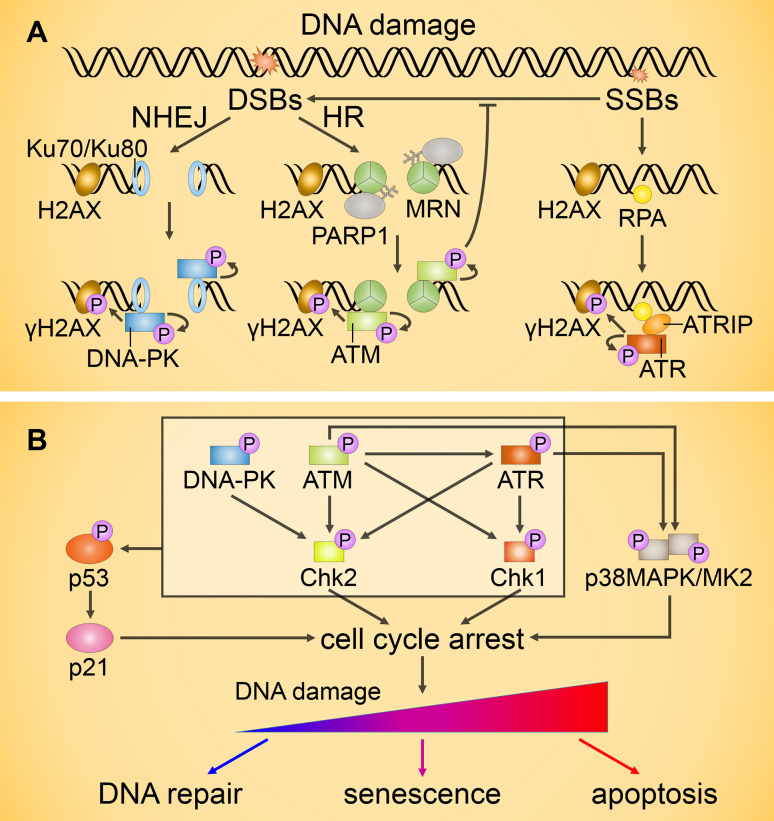
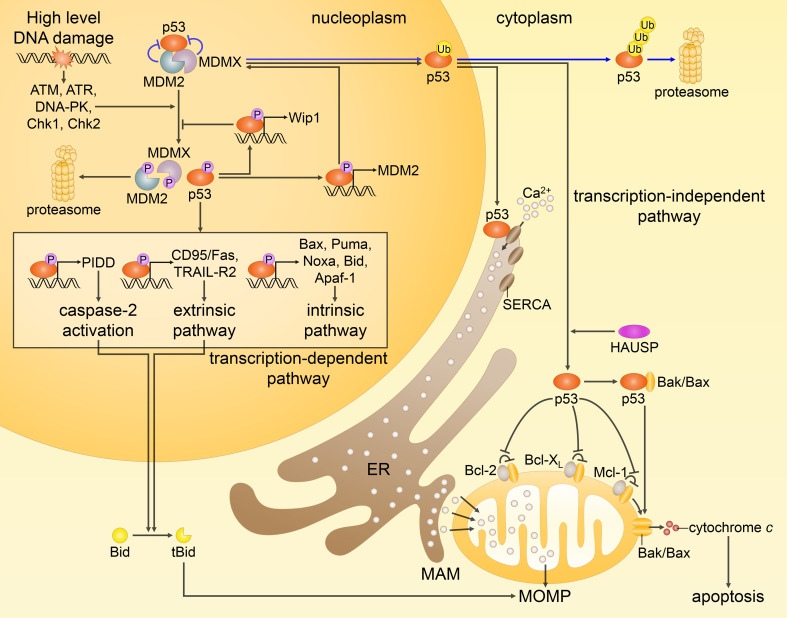
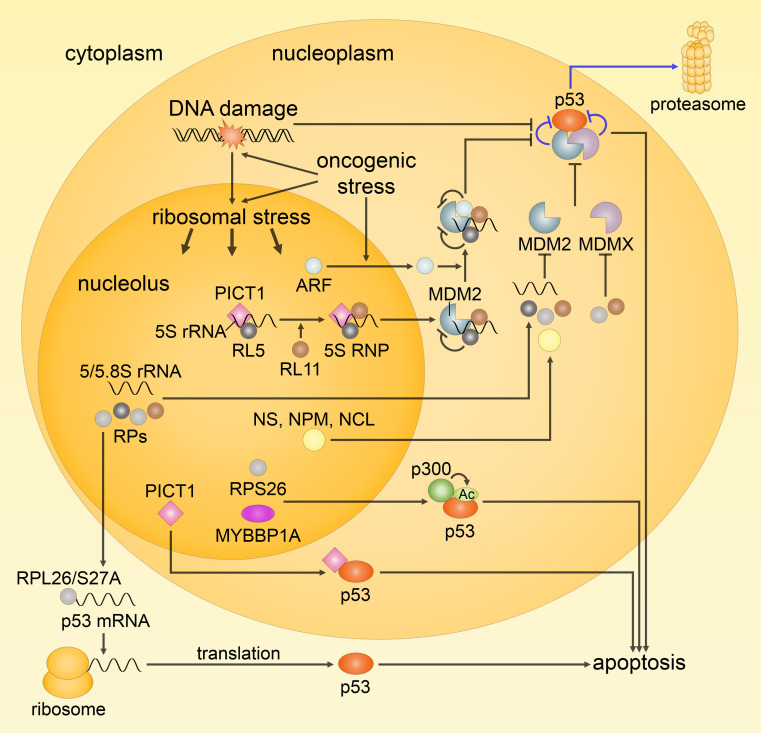
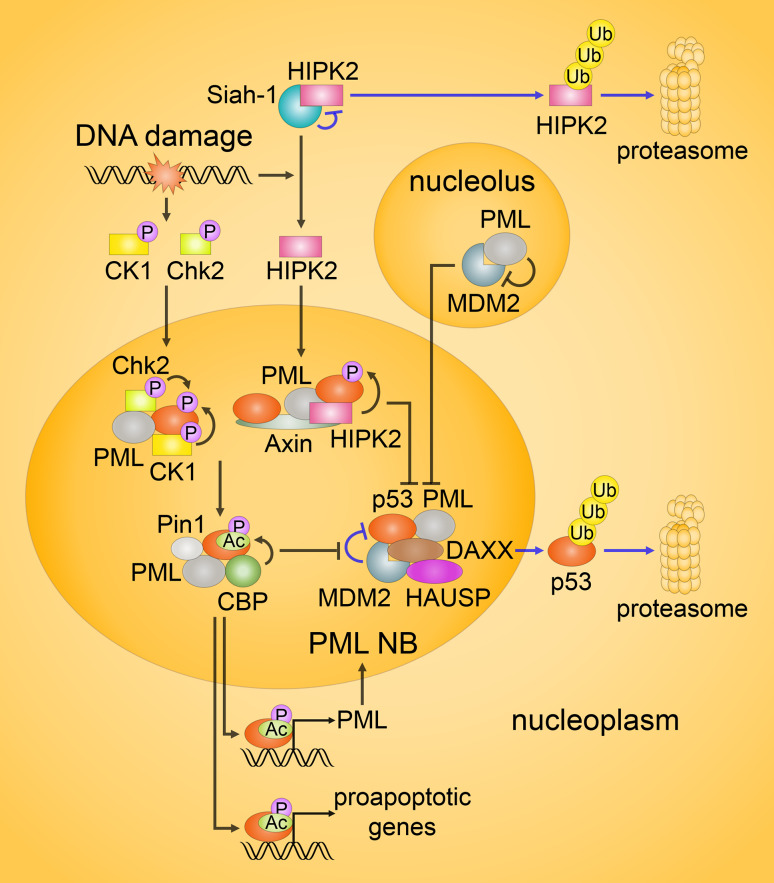
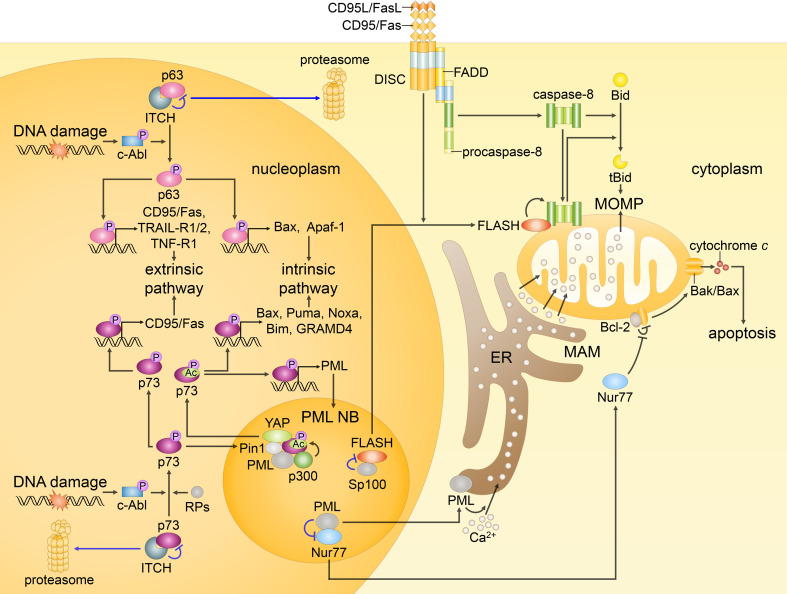
Since their establishment in the early 1970s, the nuclear changes upon apoptosis induction, such as the condensation of chromatin, disassembly of nuclear scaffold proteins and degradation of DNA, were, and still are, considered as the essential steps and hallmarks of apoptosis. These are the characteristics of the execution phase of apoptotic cell death. In addition, accumulating data clearly show that some nuclear events can lead to the induction of apoptosis. In particular, if DNA lesions resulting from deregulation during the cell cycle or DNA damage induced by chemotherapeutic drugs or viral infection cannot be efficiently eliminated, apoptotic mechanisms, which enable cellular transformation to be avoided, are activated in the nucleus. The functional heterogeneity of the nuclear organization allows the tight regulation of these signaling events that involve the movement of various nuclear proteins to other intracellular compartments (and vice versa) to initiate and govern apoptosis. Here, we discuss how these events are coordinated to execute apoptotic cell death.
Keywords: Caspases; Endonucleases; PML nuclear bodies; Ribosomal stress; p53; p63; p73.
Figures
Similar articles
-
Role of nuclear bodies in apoptosis signalling.Biochim Biophys Acta. 2008 Nov;1783(11):2185-94. doi: 10.1016/j.bbamcr.2008.07.002. Epub 2008 Jul 16. Biochim Biophys Acta. 2008. PMID: 18680765 Review.
-
Chromatin collapse during caspase-dependent apoptotic cell death requires DNA fragmentation factor, 40-kDa subunit-/caspase-activated deoxyribonuclease-mediated 3'-OH single-strand DNA breaks.J Biol Chem. 2013 Mar 29;288(13):9200-15. doi: 10.1074/jbc.M112.411371. Epub 2013 Feb 21. J Biol Chem. 2013. PMID: 23430749 Free PMC article.
-
Is cisplatin-induced cell death always produced by apoptosis?Mol Pharmacol. 2001 Apr;59(4):657-63. doi: 10.1124/mol.59.4.657. Mol Pharmacol. 2001. PMID: 11259608 Review.
-
Targeting promyelocytic leukemia protein: a means to regulating PML nuclear bodies.Int J Biol Sci. 2009 May 22;5(4):366-76. doi: 10.7150/ijbs.5.366. Int J Biol Sci. 2009. PMID: 19471587 Free PMC article. Review.
-
PML nuclear bodies and chromatin dynamics: catch me if you can!Nucleic Acids Res. 2020 Dec 2;48(21):11890-11912. doi: 10.1093/nar/gkaa828. Nucleic Acids Res. 2020. PMID: 33068409 Free PMC article. Review.
Cited by
-
The Phenomenon of Compensatory Cell Proliferation in Olfactory Epithelium in Fish Caused by Prolonged Exposure to Natural Odorants.Sci Rep. 2020 Jun 1;10(1):8908. doi: 10.1038/s41598-020-65854-9. Sci Rep. 2020. PMID: 32483178 Free PMC article.
-
Helichrysetin Induces DNA Damage that Triggers JNK-Mediated Apoptosis in Ca Ski Cells.Pharmacogn Mag. 2017 Oct-Dec;13(52):607-612. doi: 10.4103/pm.pm_53_17. Epub 2017 Nov 13. Pharmacogn Mag. 2017. PMID: 29200721 Free PMC article.
-
Diepoxybutane-induced apoptosis is mediated through the ERK1/2 pathway.Hum Exp Toxicol. 2018 Oct;37(10):1080-1091. doi: 10.1177/0960327118755255. Epub 2018 Feb 6. Hum Exp Toxicol. 2018. PMID: 29405768 Free PMC article.
-
Histone modifications as regulators of life and death in Saccharomyces cerevisiae.Microb Cell. 2015 Dec 31;3(1):1-13. doi: 10.15698/mic2016.01.472. Microb Cell. 2015. PMID: 28357312 Free PMC article. Review.
-
Orchestration of Force Generation and Nuclear Collapse in Apoptotic Cells.Int J Mol Sci. 2021 Sep 23;22(19):10257. doi: 10.3390/ijms221910257. Int J Mol Sci. 2021. PMID: 34638598 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
