

Phenotypic extremes in rare variant study designs
- PMID: 26350511
- PMCID: PMC4867440
- DOI: 10.1038/ejhg.2015.197
Phenotypic extremes in rare variant study designs
Abstract
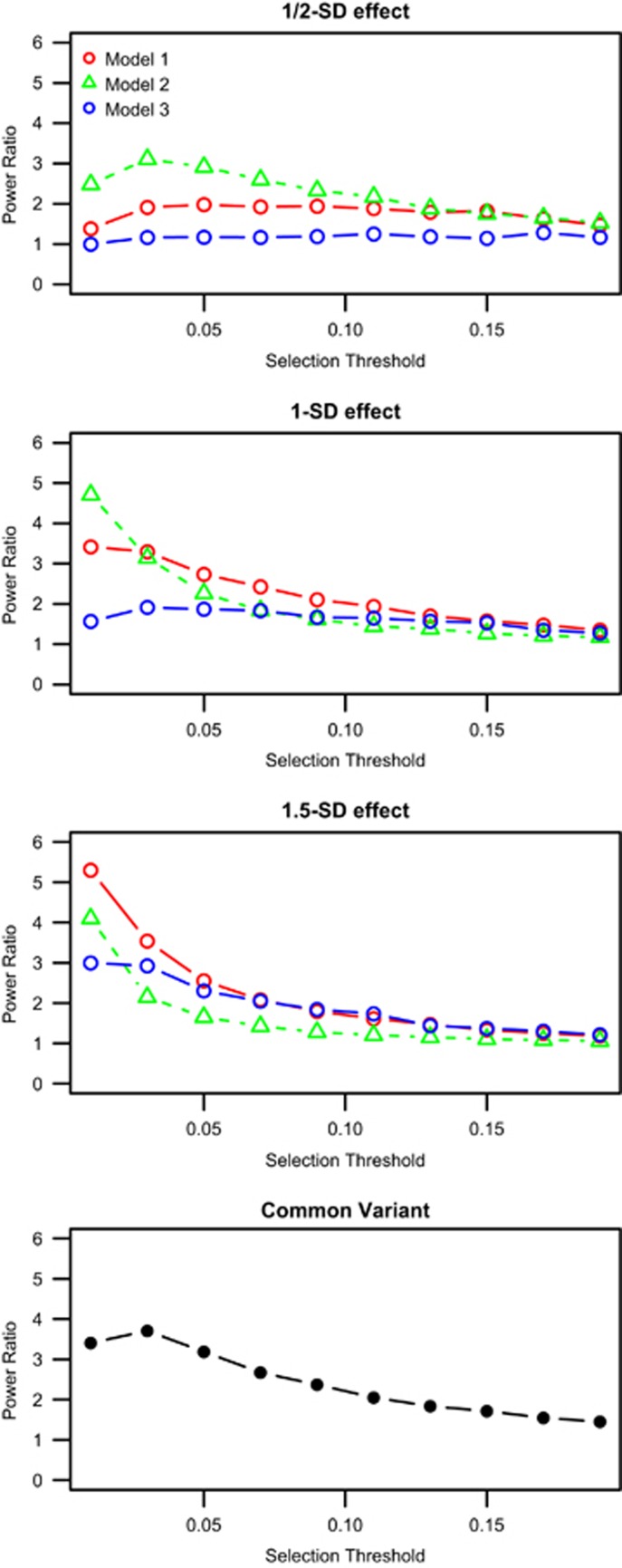
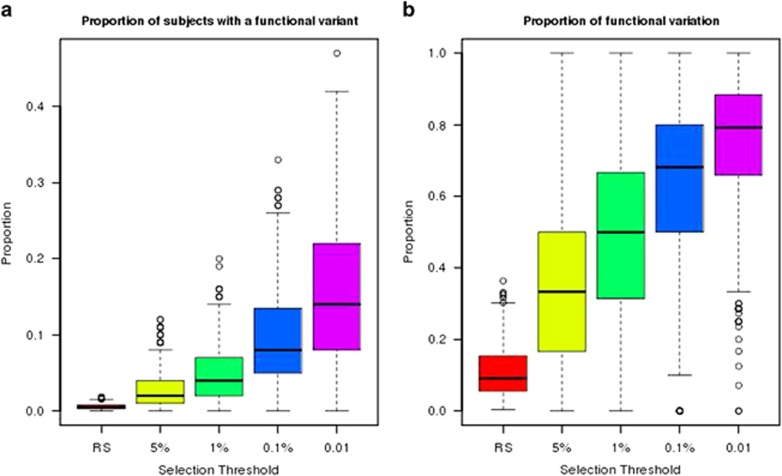
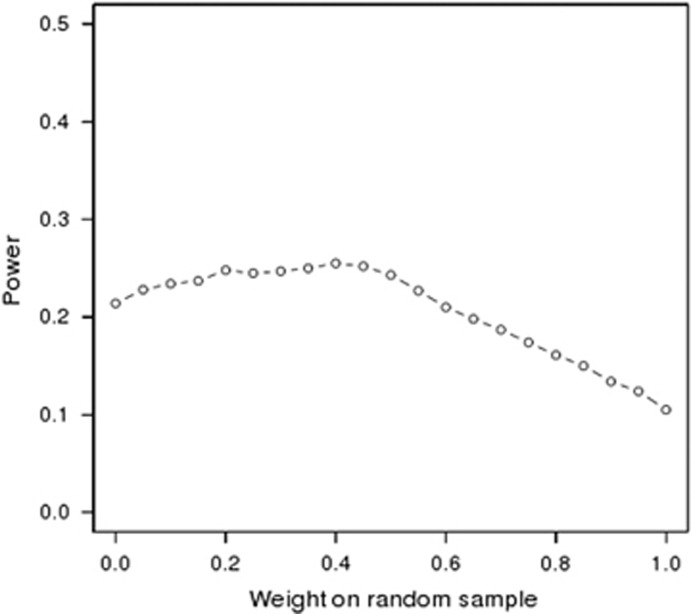
Currently, next-generation sequencing studies aim to identify rare and low-frequency variation that may contribute to disease. For a given effect size, as the allele frequency decreases, the power to detect genes or variants of interest also decreases. Although many methods have been proposed for the analysis of such data, study design and analytic issues still persist in data interpretation. In this study we present sequencing data for ABCA1 that has known rare variants associated with high-density lipoprotein cholesterol (HDL-C). We contrast empirical findings from two study designs: a phenotypic extreme sample and a population-based random sample. We found differing strengths of association with HDL-C across the two study designs (P=0.0006 with n=701 phenotypic extremes vs P=0.03 with n=1600 randomly sampled individuals). To explore this apparent difference in evidence for association, we performed a simulation study focused on the impact of phenotypic selection on power. We demonstrate that the power gain for an extreme phenotypic selection study design is much greater in rare variant studies than for studies of common variants. Our study confirms that studying phenotypic extremes is critical in rare variant studies because it boosts power in two ways: the typical increases from extreme sampling and increasing the proportion of relevant functional variants ascertained and thereby tested for association. Furthermore, we show that when combining statistical evidence through meta-analysis from an extreme-selected sample and a second separate population-based random sample, power is lower when a traditional sample size weighting is used compared with weighting by the noncentrality parameter.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
