

Combined Pan-RAF and MEK Inhibition Overcomes Multiple Resistance Mechanisms to Selective RAF Inhibitors
- PMID: 26351322
- PMCID: PMC4674359
- DOI: 10.1158/1535-7163.MCT-15-0136-T
Combined Pan-RAF and MEK Inhibition Overcomes Multiple Resistance Mechanisms to Selective RAF Inhibitors
Abstract
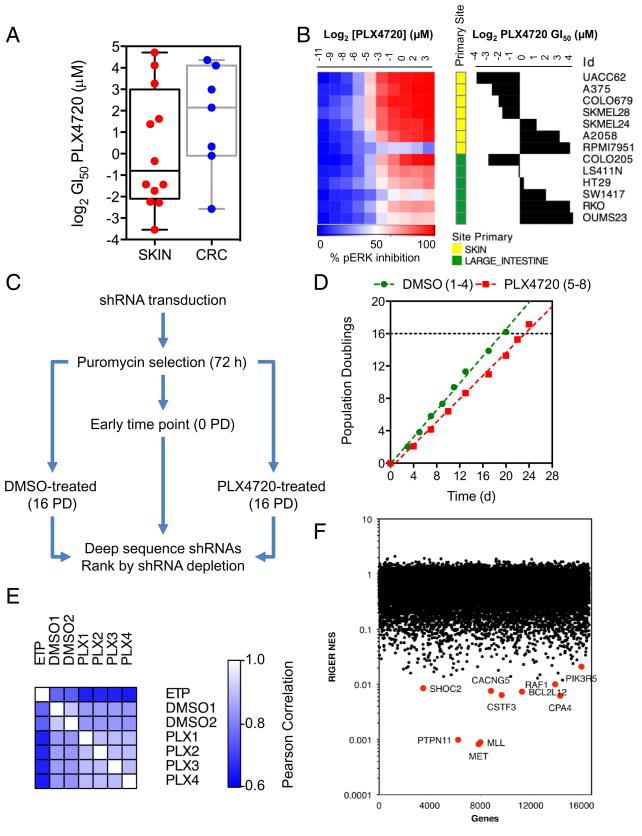
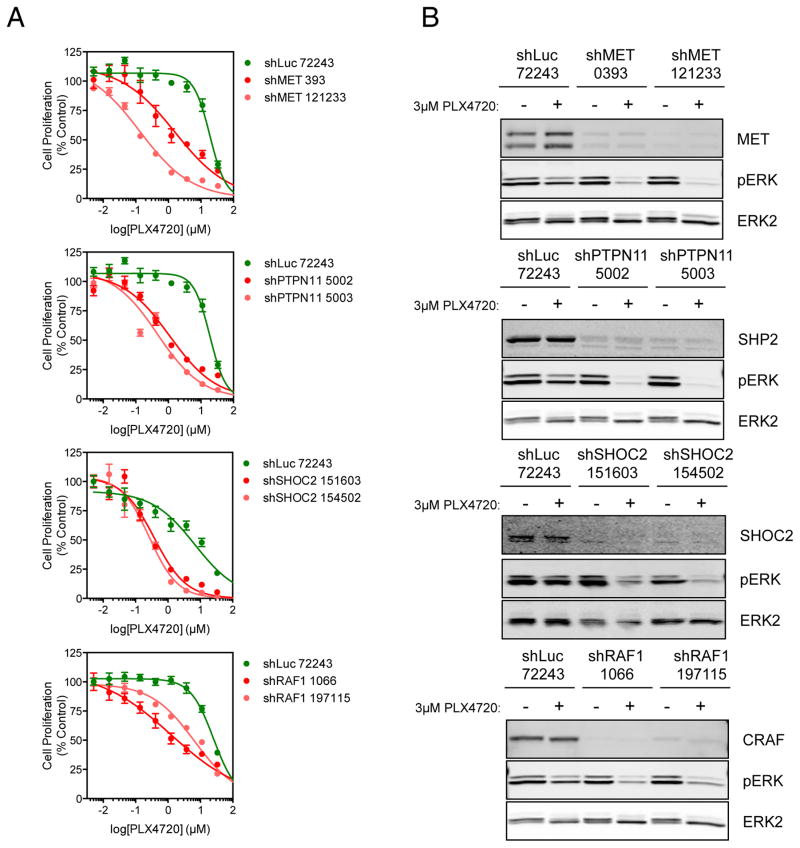
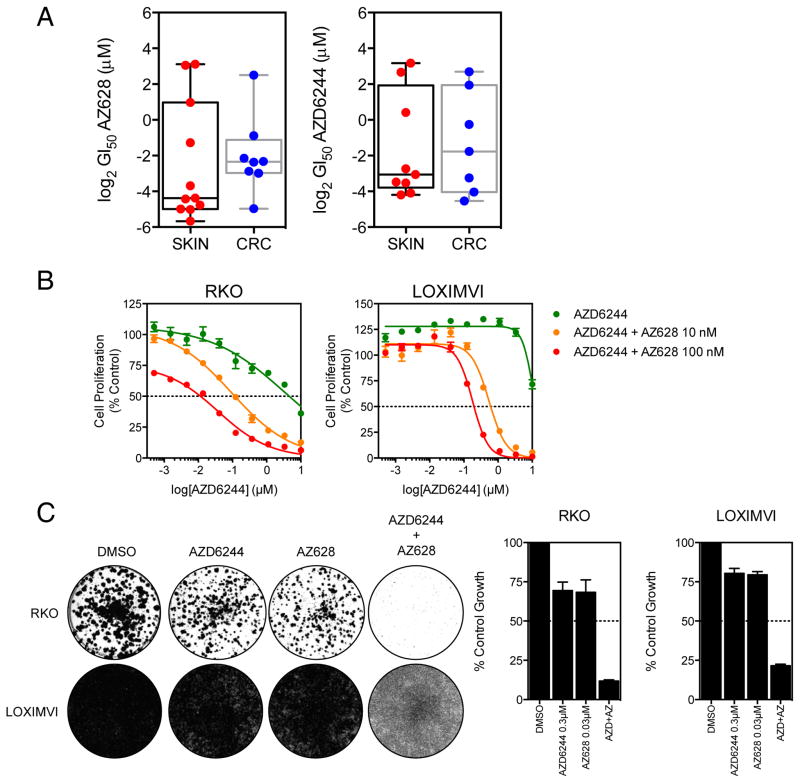
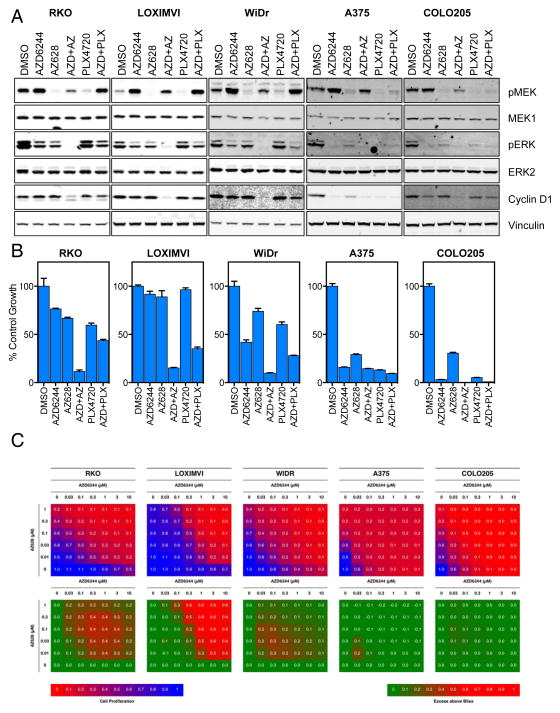
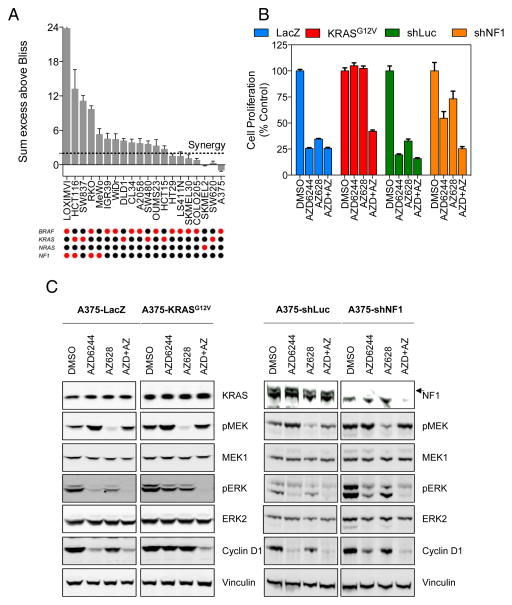
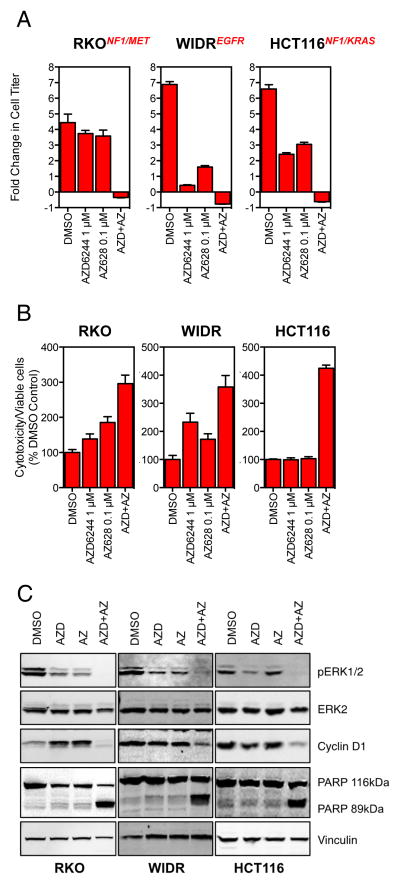
RAF and MEK inhibitors are effective in BRAF-mutant melanoma but not in BRAF-mutant colorectal cancer. To gain additional insights into this difference, we performed a genome-scale pooled shRNA enhancer screen in a BRAF-mutant, RAF inhibitor-resistant colorectal cancer cell line exposed to the selective RAF inhibitor PLX4720. We identified multiple genes along the receptor tyrosine kinase (RTK)/mitogen-activated protein kinase (MAPK) signaling axis that, when suppressed, either genetically or pharmacologically, sensitized cells to the selective RAF inhibitor through sustained inhibition of MAPK signaling. Strikingly, CRAF was a key mediator of resistance that could be overcome by the use of pan-RAF inhibitors in combination with a MEK inhibitor. Furthermore, the combination of pan-RAF and MEK inhibitors displayed strong synergy in melanoma and colorectal cancer cell lines with RAS-activating events such as RTK activation, KRAS mutation, or NF1 loss-of-function mutations. Combinations of selective RAF inhibitors, such as PLX4720 or dabrafenib, with MEK inhibitors did not incur such profound synergy, suggesting that inhibition of CRAF by pan-RAF inhibitors plays a key role in determining cellular response. Importantly, in contrast to the modest activity seen with single-agent treatment, dual pan-RAF and MEK inhibition results in the induction of apoptosis, greatly enhancing efficacy. Notably, combined pan-RAF and MEK inhibition can overcome intrinsic and acquired resistance to single-agent RAF/MEK inhibition, supporting dual pan-RAF and MEK inhibition as a novel therapeutic strategy for BRAF- and KRAS-mutant cancers.
©2015 American Association for Cancer Research.
Conflict of interest statement
Dr. Garraway is a consultant to Novartis, Millenium/Takeda, and Boehringer Ingelheim, and a recipient of a grant from Novartis.
Figures
Similar articles
-
Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations.Cancer Discov. 2015 Apr;5(4):358-67. doi: 10.1158/2159-8290.CD-14-1518. Epub 2015 Feb 11. Cancer Discov. 2015. PMID: 25673644 Free PMC article.
-
Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer.Mol Cancer Res. 2018 Mar;16(3):378-389. doi: 10.1158/1541-7786.MCR-17-0404. Epub 2017 Dec 12. Mol Cancer Res. 2018. PMID: 29233910
-
Therapeutic potential of combined BRAF/MEK blockade in BRAF-wild type preclinical tumor models.J Exp Clin Cancer Res. 2018 Jul 9;37(1):140. doi: 10.1186/s13046-018-0820-5. J Exp Clin Cancer Res. 2018. PMID: 29986755 Free PMC article.
-
Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers.Int J Mol Sci. 2015 Sep 23;16(9):22976-88. doi: 10.3390/ijms160922976. Int J Mol Sci. 2015. PMID: 26404261 Free PMC article. Review.
-
Novel mechanisms and therapeutic approaches in melanoma: targeting the MAPK pathway.Discov Med. 2015 Jun;19(107):455-61. Discov Med. 2015. PMID: 26175403 Review.
Cited by
-
Computational Modeling of Drug Response Identifies Mutant-Specific Constraints for Dosing panRAF and MEK Inhibitors in Melanoma.Cancers (Basel). 2024 Aug 22;16(16):2914. doi: 10.3390/cancers16162914. Cancers (Basel). 2024. PMID: 39199684 Free PMC article.
-
Computational modeling of drug response identifies mutant-specific constraints for dosing panRAF and MEK inhibitors in melanoma.bioRxiv [Preprint]. 2024 Aug 6:2024.08.02.606432. doi: 10.1101/2024.08.02.606432. bioRxiv. 2024. Update in: Cancers (Basel). 2024 Aug 22;16(16):2914. doi: 10.3390/cancers16162914. PMID: 39149377 Free PMC article. Updated. Preprint.
-
Drug resistance in targeted cancer therapies with RAF inhibitors.Cancer Drug Resist. 2021 Jun 17;4(3):665-683. doi: 10.20517/cdr.2021.36. eCollection 2021. Cancer Drug Resist. 2021. PMID: 35582307 Free PMC article. Review.
-
Current State of Targeted Therapy in Adult Langerhans Cell Histiocytosis and Erdheim-Chester Disease.Target Oncol. 2024 Sep;19(5):691-703. doi: 10.1007/s11523-024-01080-x. Epub 2024 Jul 11. Target Oncol. 2024. PMID: 38990463 Review.
-
Cosmosiin Induces Apoptosis in Colorectal Cancer by Inhibiting PD-L1 Expression and Inducing ROS.Antioxidants (Basel). 2023 Dec 18;12(12):2131. doi: 10.3390/antiox12122131. Antioxidants (Basel). 2023. PMID: 38136250 Free PMC article.
References
-
- Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. ASCO Meeting Abstracts. 2010;28:3534.
-
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12372–7. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
