

Gut DNA viromes of Malawian twins discordant for severe acute malnutrition
- PMID: 26351661
- PMCID: PMC4586842
- DOI: 10.1073/pnas.1514285112
Gut DNA viromes of Malawian twins discordant for severe acute malnutrition
Abstract
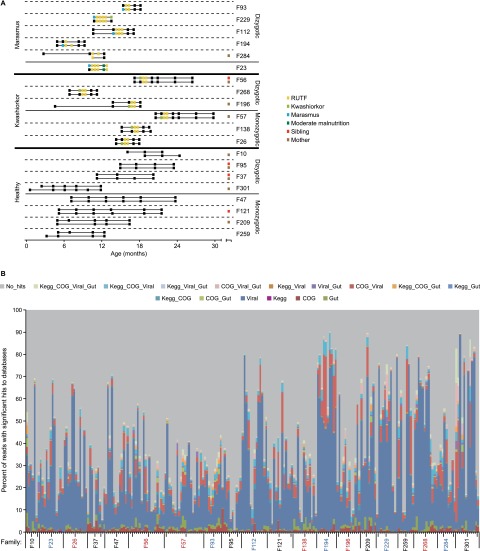
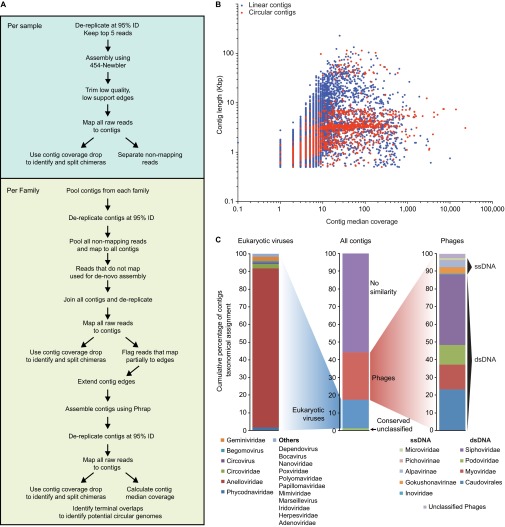
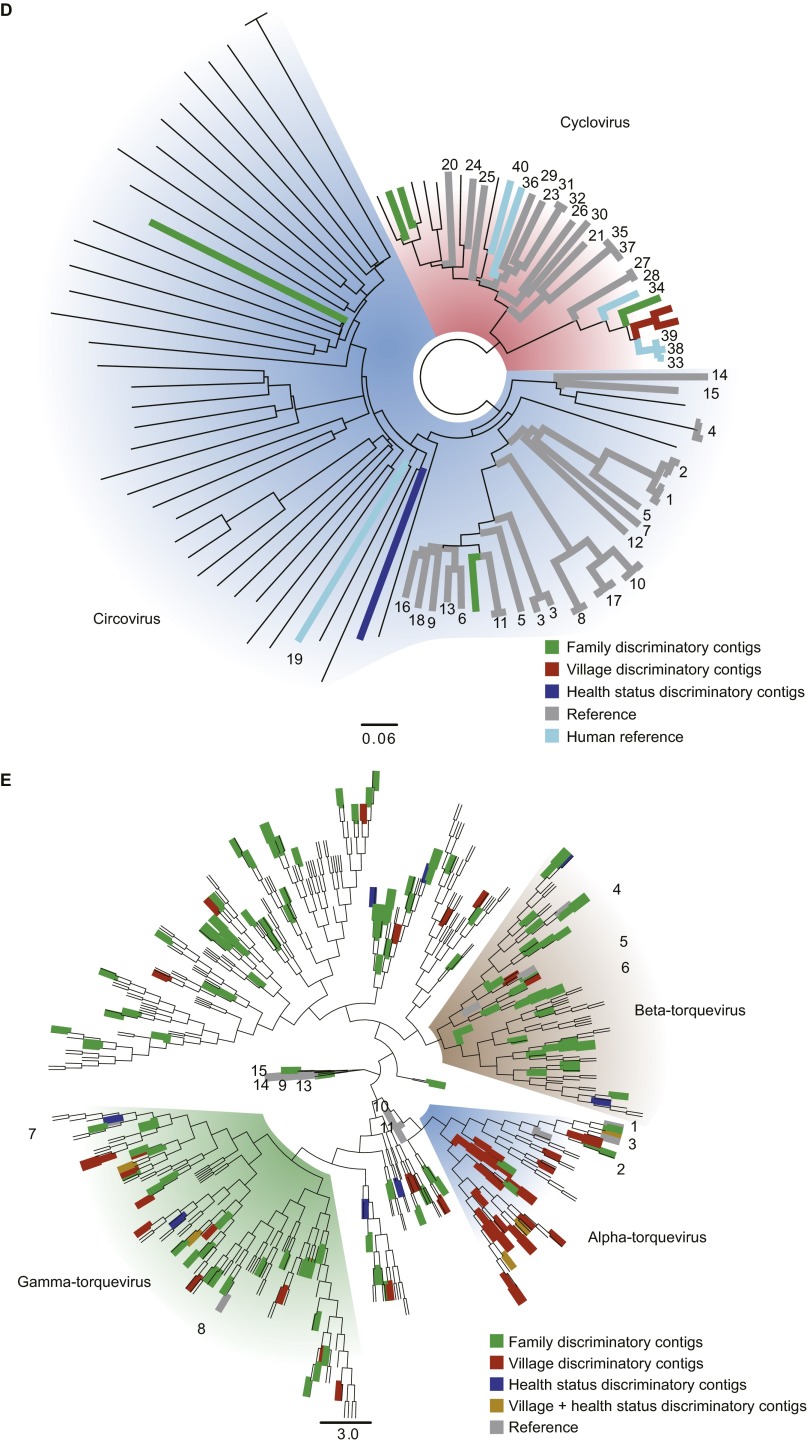
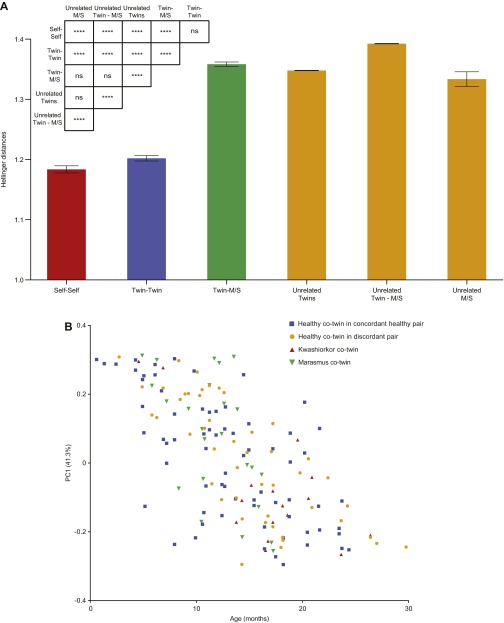
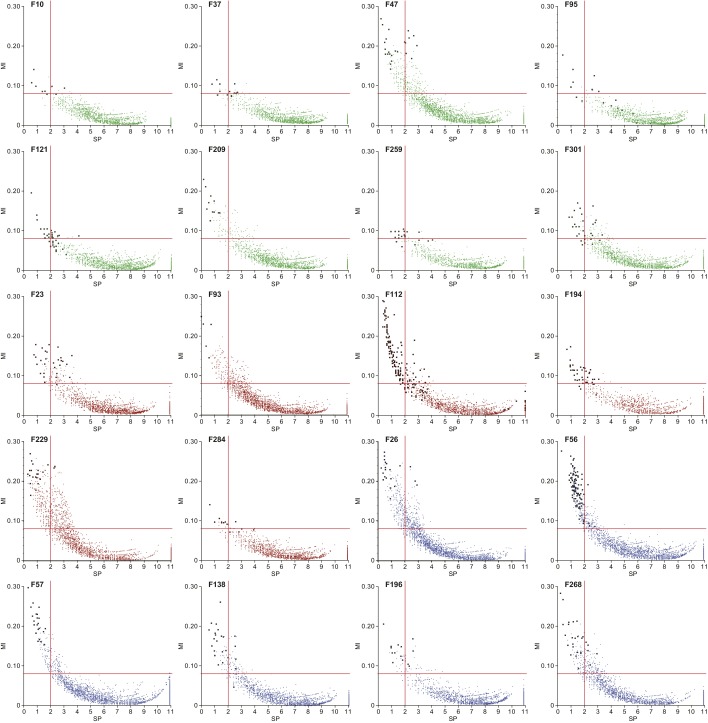
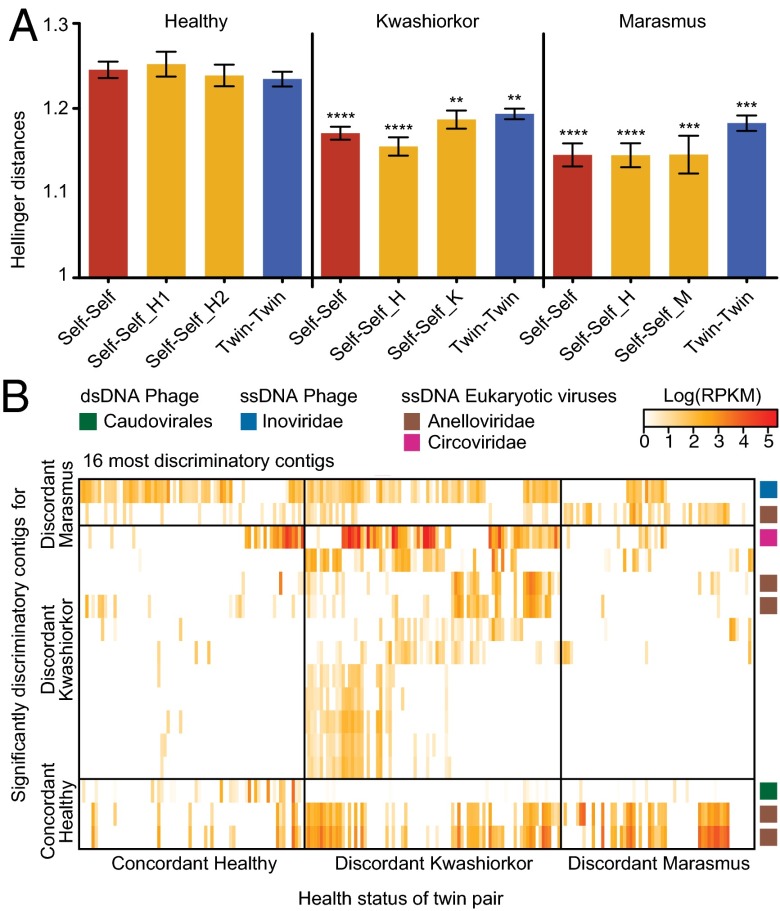
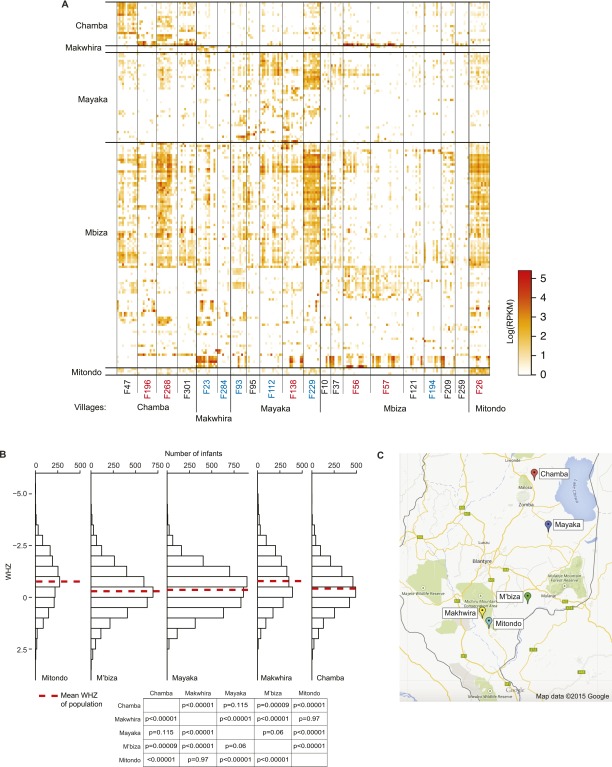
The bacterial component of the human gut microbiota undergoes a definable program of postnatal development. Evidence is accumulating that this program is disrupted in children with severe acute malnutrition (SAM) and that their persistent gut microbiota immaturity, which is not durably repaired with current ready-to-use therapeutic food (RUTF) interventions, is causally related to disease pathogenesis. To further characterize gut microbial community development in healthy versus malnourished infants/children, we performed a time-series metagenomic study of DNA isolated from virus-like particles (VLPs) recovered from fecal samples collected during the first 30 mo of postnatal life from eight pairs of mono- and dizygotic Malawian twins concordant for healthy growth and 12 twin pairs discordant for SAM. Both members of discordant pairs were sampled just before, during, and after treatment with a peanut-based RUTF. Using Random Forests and a dataset of 17,676 viral contigs assembled from shotgun sequencing reads of VLP DNAs, we identified viruses that distinguish different stages in the assembly of the gut microbiota in the concordant healthy twin pairs. This developmental program is impaired in both members of SAM discordant pairs and not repaired with RUTF. Phage plus members of the Anelloviridae and Circoviridae families of eukaryotic viruses discriminate discordant from concordant healthy pairs. These results disclose that apparently healthy cotwins in discordant pairs have viromes associated with, although not necessarily mediators, of SAM; as such, they provide a human model for delineating normal versus perturbed postnatal acquisition and retention of the gut microbiota's viral component in populations at risk for malnutrition.
Keywords: age/disease-discriminatory phage and eukaryotic viruses; assembly of the human gut DNA virome; childhood malnutrition; epidemiology; gnotobiotic mice.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
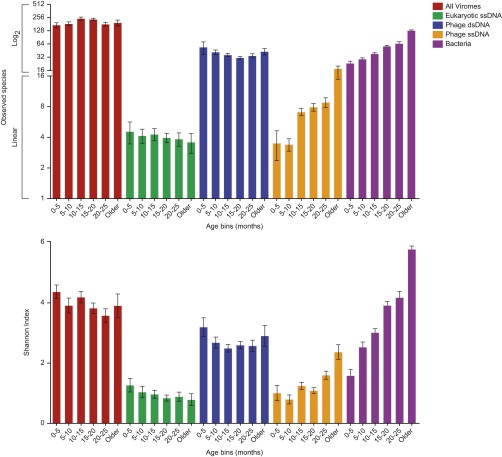
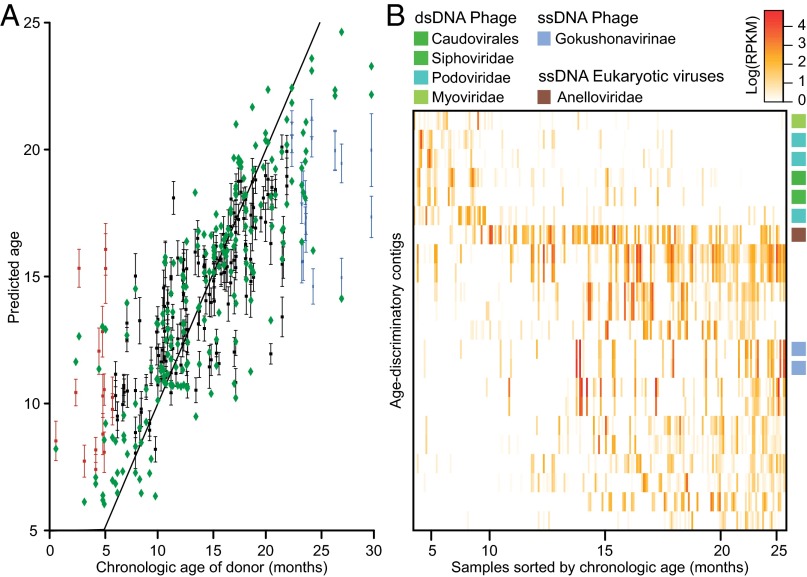
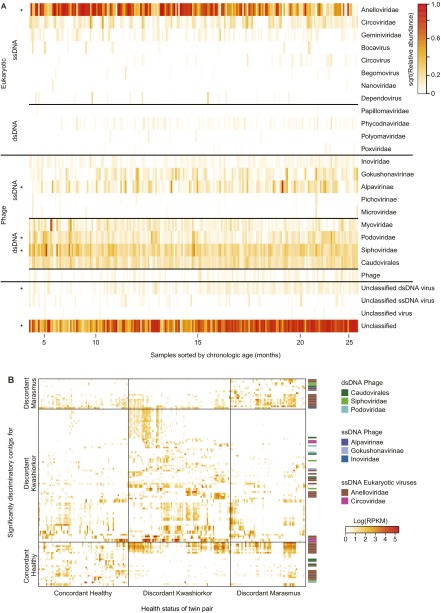
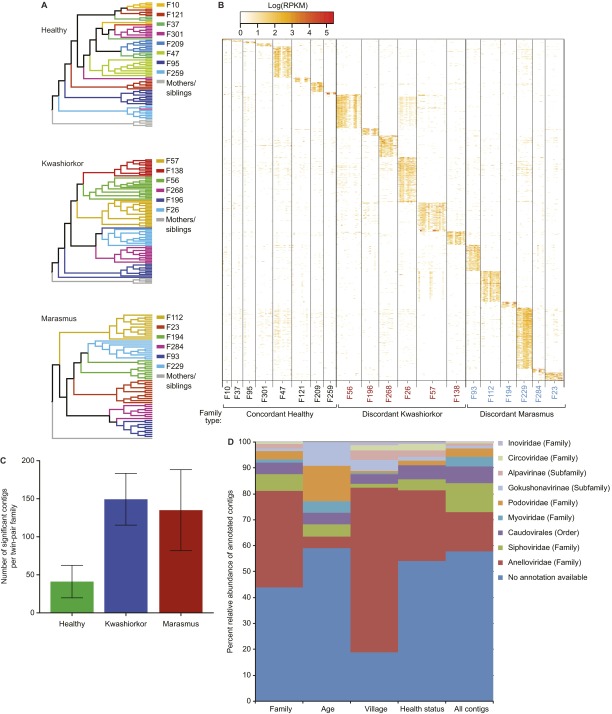
References
-
- Black RE, et al. Maternal and Child Nutrition Study Group Maternal and child undernutrition and overweight in low-income and middle-income countries. Lancet. 2013;382(9890):427–451. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
